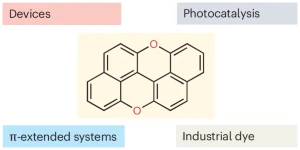
Davide Zanetti, Cristian De Luca, and Davide Bonifazi discuss the resurgence of peri-xanthenoxanthene from a forgotten dye into a versatile organic semiconductor and sustainable photocatalyst, emphasizing its importance in photoredox catalysis.
Publications
2025
Title
A bright comeback for peri-xanthenoxanthene
Davide Zanetti, Cristian De Luca, Davide Bonifazi
Journal
Nature Chemistry
Date
07/2025
Nature Chemistry
07/2025
A bright comeback for peri-xanthenoxanthene
3 Jul 2025
Abstract
Bottom-up fabrication of BN-doped graphene electrodes from thiol-terminated borazine molecules working in solar cells
Carolina M Ibarra-Barreno, Sanchari Chowdhury, Martina Crosta, Tashfeen Zehra, Francesco Fasano, Paromita Kundu, Jenthe Verstraelen, Sara Bals, Mohammed Subrati, Davide Bonifazi, Rubén D Costa, Petra Rudolf
Journal
ACS Applied Materials & Interfaces
Date
04/2025
ACS Applied Materials & Interfaces
04/2025
Bottom-up fabrication of BN-doped graphene electrodes from thiol-terminated borazine molecules working in solar cells
Carolina M Ibarra-Barreno, Sanchari Chowdhury, Martina Crosta, Tashfeen Zehra, Francesco Fasano, Paromita Kundu, Jenthe Verstraelen, Sara Bals, Mohammed Subrati, Davide Bonifazi, Rubén D Costa, Petra Rudolf
ACS Applied Materials & Interfaces
2 Apr 2025
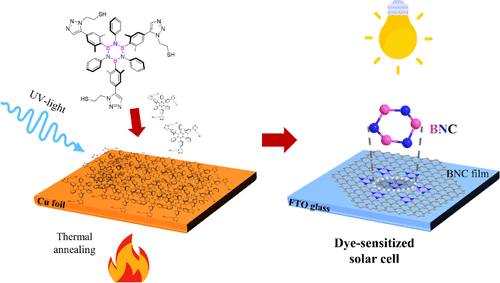
Abstract
Graphene exhibits exceptional properties, including high tensile strength, mechanical stiffness, and electron mobility. Chemical functionalization of graphene with boron and nitrogen is a powerful strategy for tuning these properties for specific applications. Molecular self-assembly provides an efficient pathway for the tailored synthesis of doped graphene, depending on the molecular precursor used. This study presents a scalable approach to synthesizing large-area boron- and nitrogen-doped graphene using two borazine precursors bearing thiol functionalities. After self-assembly on electropolished polycrystalline copper foil, the precursors undergo photopolymerization under UV irradiation, and subsequent annealing in vacuum transforms the cross-linked BN-doped layer into a graphenoid structure. X-ray photoelectron spectroscopy confirms the integration of the borazine rings into the BNC architecture, while Raman spectroscopy reveals a red shift in the characteristic G bands along with intense and broad D bands, highlighting boron–nitrogen contributions. Transmission electron microscopy provides insight into the morphology and structural quality of the BNC films. The BNC films were successfully integrated as counter electrodes in dye-sensitized solar cells, achieving a power conversion efficiency of up to 6% under 1 sun illumination and 11.8% under low-intensity indoor ambient light. Hence, this work not only establishes a straightforward, controllable route for heteroatom doping but also introduces a novel concept of Pt-free counter electrodes for efficient indoor energy harvesting applications.
Catalyst-Transfer Macrocyclization Protocol: Synthesis of π-Conjugated Azaparacyclophanes Made Easy
Josue Ayuso-Carrillo, Davide Bonifazi
Journal
JACS Au
Date
03/2025
JACS Au
03/2025
Catalyst-Transfer Macrocyclization Protocol: Synthesis of π-Conjugated Azaparacyclophanes Made Easy
7 Mar 2025

Abstract
The present Protocol describes the application of the catalyst-transfer macrocyclization (CTM) reaction, focusing on the synthesis of aza[1n]paracyclophanes (APCs). APCs are fully π-conjugated shape-persistent macrocycles with potential supramolecular chemistry and materials science applications. This method leverages the Pd-catalyzed Buchwald–Hartwig cross-coupling reaction to selectively form π-conjugated cyclic structures, a significant advancement due to its efficiency, versatility, and scalability. Overall, this Article highlights the following attributes of the CTM method: a) Efficiency and Yield: The CTM method works at mild temperatures (40 °C) and short reaction times (≥2 h), producing high yields of APCs (>75% macrocycles). It avoids the typical high-dilution conditions, making it more practical for large-scale applications. b) Versatility: The method allows the synthesis of APCs with diverse endocyclic and exocyclic functionalities and ring sizes (typically from 4- to 9-membered rings), expanding the chemical space for these compounds. This flexibility is crucial for tailoring APC properties for specific applications. c) Scalability and Reproducibility: Unlike many macrocyclization reactions, which require highly dilute conditions, CTM can perform under concentrated regimes (35–350 mM), making it more suitable for large-scale applications. d) Applications in Materials Science: APCs are noted for their potential in optoelectronic applications due to their π-conjugated structures, which are helpful in organic semiconductors, light-harvesting systems, and other advanced materials. This approach addresses the challenge of complicated multistep syntheses that have hindered the widespread integration of APCs into functional devices. A step-by-step guide to preparing exemplary APCs, including troubleshooting, is provided with photographic illustrations.
Combining broadband absorbing electrochromic materials for hybrid grey-to-colourless flexible devices
Lukas Niklaus, Rúben R Ferreira, Sven Macher, Antoine Stopin, Marco Schott, Laura Maggini, Davide Bonifazi
DOI: 10.1039/D4CC06804A
Journal
Chemical Communications
Date
03/2025
Chemical Communications
03/2025
Combining broadband absorbing electrochromic materials for hybrid grey-to-colourless flexible devices
Lukas Niklaus, Rúben R Ferreira, Sven Macher, Antoine Stopin, Marco Schott, Laura Maggini, Davide Bonifazi
DOI: 10.1039/D4CC06804A
Chemical Communications
6 Mar 2025
Abstract
This study presents a grey-to-colourless hybrid electrochromic device combining a novel red-to-colourless polymer with blue-to-colourless Prussian blue. The device achieves a neutral tint in both the dark and bleached states, with notable changes in visible light transmittance and fast response.
Cyano‐Borazine Photosensitizers for Dye‐Sensitized Solar Cells
Sanchari Chowdhury, Vivek Chandrakant Wakchure, El Czar Galleposo, Davide Bonifazi, Rubén D Costa
Journal
Advanced Energy and Sustainability Research
Date
01/2025
Advanced Energy and Sustainability Research
01/2025
Cyano‐Borazine Photosensitizers for Dye‐Sensitized Solar Cells
Sanchari Chowdhury, Vivek Chandrakant Wakchure, El Czar Galleposo, Davide Bonifazi, Rubén D Costa
Advanced Energy and Sustainability Research
13 Jan 2025
Abstract
Implementing novel metal-free and strongly absorbing donor–acceptorsensitizers without carboxylic acid anchoring groups are still a frontier in dye-sensitized solar cells (DSSCs). Herein, the facile synthesis of a strongly absorbingsensitizer combining three 1,1,4,4-tetracyanobuta-1,3-diene (TCBD) anchoringmoieties with a borazine core instead of the classical cyano anchoring groups,such as tetracyanoquinodimethane (TCNQ) and tetracyanoethylene (TCNE), andthe dimethyl-phenyl amino donor group, is disclosed. This results in a 1.6-foldincrease in solar energy conversion efficiency compared to DSSCs with thereference sensitizers (TCBD-dimethyl-amino-phenyl core) and the prior art cyano-sensitizers with TCNE and TCNG anchors. The advantages of the TCBD-borazinedesign are twofold: 1) threefold increase in absorption extinction coefficient aswell as 2) a reduction in back electron transfer and aggregation behavior upon dyeadsorption onto the semiconducting electrode, resulting in 45% and 23%improvement in open-circuit voltage (V oc) and short-circuit current density (J sc ),respectively, compared to those of the prior art. Overall, this work highlights aneasy-to-design of cyano-sensitizer that results in a significant improvement ofsolar energy conversion when using borazine frameworks for the first time.
2024
Title
Assessing the energetic and environmental sustainability of organic borazines preparation: A comprehensive life cycle assessment and uncertainty analysis
Filippo Campana, Kejie Zhou, Jhonny A Yunda, Alireza Nazari Khodadadi, Davide Bonifazi, Sorin Melinte, Luigi Vaccaro
Journal
Chemical Engineering Journal
Date
12/2024
Chemical Engineering Journal
12/2024
Assessing the energetic and environmental sustainability of organic borazines preparation: A comprehensive life cycle assessment and uncertainty analysis
Filippo Campana, Kejie Zhou, Jhonny A Yunda, Alireza Nazari Khodadadi, Davide Bonifazi, Sorin Melinte, Luigi Vaccaro
Chemical Engineering Journal
20 Dec 2024
Abstract
Since its inception, organic synthesis has played a fundamental role in the development of society, as its efficiency is essential for the preparation of materials in several strategic sectors such as pharmaceuticals, transportation and energy. In this context, organic borazines have emerged as promising molecules useful both as doping units and organic semiconductors, particularly in the production of photovoltaics and organic transistors. However, like most “fine chemical” products, their engineering is generally complex and harmful to the environment due to the need for dangerous reagents, solvents, and harsh reaction conditions. Recent adopted advancements in the manufacturing process, including continuous-flow synthesis and the use of safer, biomass-derived solvents, have been confirmed through a comprehensive cradle-to-gate life cycle assessment (LCA). The study, compared to four batch processes from the literature, identified electricity consumption as the primary contributor to environmental and human health impacts. Additionally, it was demonstrated that adopting a continuous-flow approach, which reduces electricity consumption and leverages safer reaction media such as 2-MeTHF, characterized by an exceptional recovery rate (90%), proved to be an effective strategy, resulting in a notable 11% reduction in emissions. Furthermore, an uncertainty analysis using the Monte Carlo method revealed that energy mixes reliant on fossil fuels increase the impacts across all categories related to human health damage.
Expression of hyperconjugative stereoelectronic interactions in borazines
Vivek Chandrakant Wakchaure, Jacopo Dosso, Martina Crosta, Hanspeter Kählig, Benjamin D Ward, Davide Bonifazi
DOI: 10.1039/d4cc05188b
Journal
Chemical Communications
Date
12/2024
Chemical Communications
12/2024
Expression of hyperconjugative stereoelectronic interactions in borazines
Vivek Chandrakant Wakchaure, Jacopo Dosso, Martina Crosta, Hanspeter Kählig, Benjamin D Ward, Davide Bonifazi
DOI: 10.1039/d4cc05188b
Chemical Communications
4 Dec 2024
Abstract
This paper discusses hyperconjugative stereoelectronic effects in borazines. A series of alkyl-substituted borazines were synthesized and analysed by NMR spectroscopy and X-ray diffraction. Supported by NBO analyses, the significant decreases in 1JCH coupling constant for the CH groups adjacent to the boron atoms are consistent with the presence of and interactions. These interactions lower the electrophilicity of boron atoms, enhancing moisture stability and establishing these molecules as valuable scaffolds in synthetic chemistry and materials science.
Escape from Flatland: Stereoselective Synthesis of Hexa-aryl Borazines and their sp²-Based 3D Architectures
Vivek C Wakchaure, Maria Mercedes Lorenzo-García, Francesco Fasano, Martina Crosta, Nicolas Biot, Pradip Kumar Mondal, Nicola Demitri, Benjamin Ward, Davide Bonifazi
Journal
Angewandte Chemie International Edition
Date
11/2024
Angewandte Chemie International Edition
11/2024
Escape from Flatland: Stereoselective Synthesis of Hexa-aryl Borazines and their sp²-Based 3D Architectures
Vivek C Wakchaure, Maria Mercedes Lorenzo-García, Francesco Fasano, Martina Crosta, Nicolas Biot, Pradip Kumar Mondal, Nicola Demitri, Benjamin Ward, Davide Bonifazi
Angewandte Chemie International Edition
12 Nov 2024
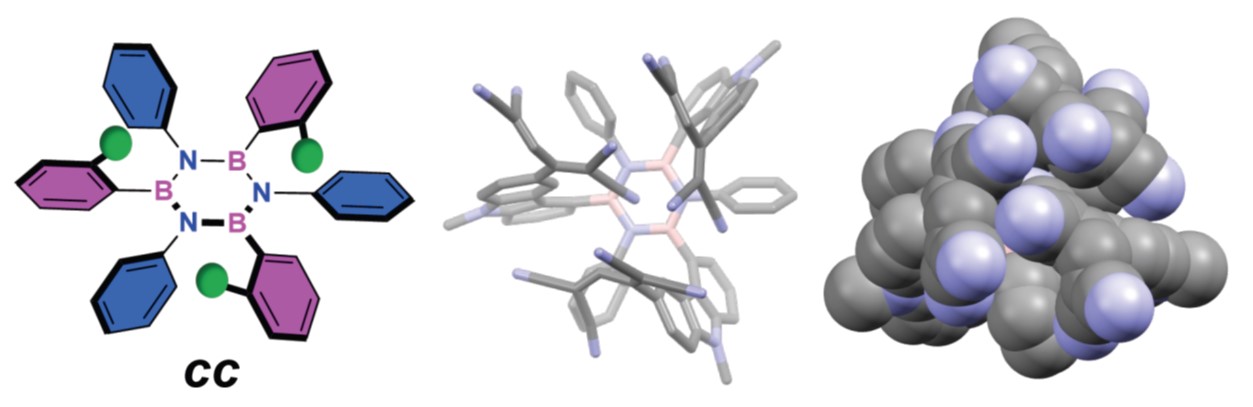
Abstract
Borazine and its derivatives can be considered critical doping units for engineering hybrid C(sp2)-based molecules with tailored optoelectronic properties. Herein, we report the first synthesis of hexaarylborazines that, bearing ortho-substituted aryl moieties, extend three-dimensionally. Using a one-pot protocol, we first form an electrophilic chloroborazole and then react it with an aryl lithium (ArLi). By selecting the appropriate ortho-substituent, we can guide the ArLi to add to the BN-core in a specific way, ultimately controlling the stereochemical outcome of the three-substitution reaction. Rationalization of the stereochemical model through computational analysis allowed us to show that when aryl lithium nucleophiles bearing rigid long-range ortho-substituents are used, i.e., stiff substituents. The ortho-substituent shields its side of the electrophilic B3N3 core, biasing the incoming ArLi to add anti at each addition step, forming the final tri-aryl borazine exclusively as cc-isomer. Leveraging this stereoselective approach, prototypical multichromophoric borazine derivatives were prepared, and we showcased how the stereochemical arrangement of these chromophores distinctly influences their redox behavior. This methodology paves the way for previously inaccessible borazines to serve as privileged precursors to transcend the conventional bidimensionality associated with graphenoid systems and pioneer the construction of new forms of three-dimensional C(sp2)-based architectures.
Long-range supramolecular assembly of a pyrene-derivatized polythiophene/MWCNT hybrid for resilient flexible electrochromic displays
Rúben R Ferreira, Dario Mosca, Tiago Moreira, Vivek Chandrakant Wakchaure, Gianvito Romano, Antoine Stopin, Carlos Pinheiro, Alexander MT Luci, Luís MA Perdigão, Giovanni Costantini, Heinz Amenitsch, Cesar AT Laia, A Jorge Parola, Laura Maggini, Davide Bonifazi
Journal
ACS Applied Engineering Materials
Date
10/2024
ACS Applied Engineering Materials
10/2024
Long-range supramolecular assembly of a pyrene-derivatized polythiophene/MWCNT hybrid for resilient flexible electrochromic displays
Rúben R Ferreira, Dario Mosca, Tiago Moreira, Vivek Chandrakant Wakchaure, Gianvito Romano, Antoine Stopin, Carlos Pinheiro, Alexander MT Luci, Luís MA Perdigão, Giovanni Costantini, Heinz Amenitsch, Cesar AT Laia, A Jorge Parola, Laura Maggini, Davide Bonifazi
ACS Applied Engineering Materials
21 Oct 2024
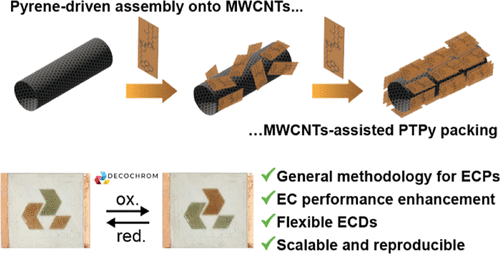
Abstract
Organic electrochromic polymers hold great potential for integration into low-power flexible electrochromic displays (F-ECDs) due to their wide range of colors and simple processing. However, challenges such as inefficient charge transfer and degradation upon device integration hinder their practical applications. Herein, we report an innovative, general approach that utilizes template-induced supramolecular nanostructuring to engineer established electrochromic polymers, enhancing their performance and durability. We modified a well-known, albeit underperforming in F-ECDs, poly-thiophene polymer (ECP Orange; PT) by incorporating a pyrene appendage, resulting in a copolymer (PTPy) capable of undergoing large-scale assembly in the presence of multi-walled carbon nanotubes (MWCNTs), driven by the establishment of π–π interactions between the pyrene and the MWCNTs (PTPy/MWCNTs). F-ECDs based on these hybrids, produced by spray coating, exhibit improved color switching speeds (t90OX = 3.6 s, t90RED = 0.3 s) compared to those of the PT polymer (t90OX = 53.2 s, t90RED = 2.5 s). Additionally, PTPy/MWCNTs F-ECDs demonstrate longer cyclability (half-life based on ΔE, ΔE50% = 17.6k cycles) compared to PT (ΔE50% = 278 cycles), also when blended with MWCNTs (ΔE50% = 282 cycles). This work highlights the pivotal role of engineered supramolecular nanostructuring in boosting the performance of organic electrochromic materials, making them suitable for F-ECD scalable commercial applications.
Hybrid Screen Printable Electrolyte for Large-Scale Flexible Electrochromic Display Production
Fábio A. S. Leite, Piotr Wierzchowiec, Carlos Pinheiro, Laura Maggini, Davide Bonifazi
Journal
Advanced Materials Technologies
Date
10/2024
Advanced Materials Technologies
10/2024
Hybrid Screen Printable Electrolyte for Large-Scale Flexible Electrochromic Display Production
Fábio A. S. Leite, Piotr Wierzchowiec, Carlos Pinheiro, Laura Maggini, Davide Bonifazi
Advanced Materials Technologies
3 Oct 2024
Abstract
This study presents the development of a novel screen printable quasi-solid polymer electrolyte (p-QSPE) for Electrochromic Displays (ECDs) applications. p-QSPE is composed of three key components: polyvinylidene fluoride (PVDF), a high-dielectric constant polymer that ensures high ionic conductivity in solid-state; glyceril propoxy triacrylate (GPTA), a UV-cross-linkable monomer that provides structure and durability for overprinting; titanium dioxide (TiO₂) nanoparticles, which modulate the electrolyte’s rheological properties for screen printing; reducing the solvent (PC:EC) content to only 35.90 wt.%. Electrochemical Impedance Spectroscopy (EIS) revealed that this well-designed formulation achieved an ionic conductivity of 1.17 × 10−3 S cm−1 at room temperature, surpassing the threshold required for commercial applications. Moreover, p-QSPE facilitated the production of fully screen printed ECDs in an industrial printing line, streamlining their production process and achieving an optimal balance between printability, overprint resilience, and device performance. Operational tests for the ECDs showed fast switching times (<6 s for t90 and <2 s for t75) across a wide temperature range (−20 °C to 80 °C). Additionally, the electrolyte demonstrated low charge consumption (2.10 ± 0.11 mC cm−2) and a lifespan exceeding 10 000 cycles. These results highlight the potential of p-QSPE as a screen printable, high-performance electrolyte, capable of advancing ECD manufacturing by enabling the production of fully screen-printed, performing ECDs.
Surface Chemistry of a Halogenated Borazine: From Supramolecular Assemblies to a Random Covalent BN‐Substituted Carbon Network
Birce Sena Tömekce, Marc G Cuxart, Laura Caputo, Daniele Poletto, Jean-Christophe Charlier, Davide Bonifazi, Willi Auwärter
Journal
Chemistry–A European Journal
Date
09/2024
Chemistry–A European Journal
09/2024
Surface Chemistry of a Halogenated Borazine: From Supramolecular Assemblies to a Random Covalent BN‐Substituted Carbon Network
Birce Sena Tömekce, Marc G Cuxart, Laura Caputo, Daniele Poletto, Jean-Christophe Charlier, Davide Bonifazi, Willi Auwärter
Chemistry–A European Journal
7 Sep 2024
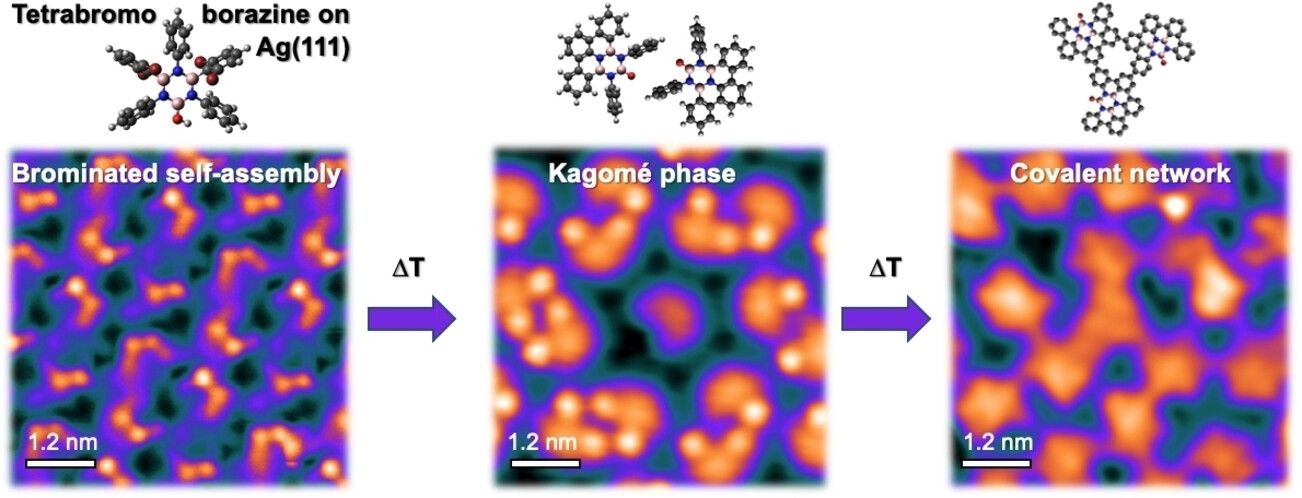
Abstract
The on‐surface synthesis strategy has emerged as a promising route for fabricating well‐defined two‐dimensional (2D) BN‐substituted carbon nanomaterials with tunable electronic properties. This approach relies on specially designed precursors and requires a thorough understanding of the on‐surface reaction pathways. It promises precise structural control at the atomic scale, thus complementing chemical vapor deposition (CVD). In this study, we investigated a novel heteroatomic precursor, tetrabromoborazine, which incorporates a BN core and an OH group, on Ag(111) using low temperature scanning tunnelling microscopy/spectroscopy (LT‐STM/STS) and X‐ray photoelectron spectroscopy (XPS). Through sequential temperature‐induced reactions involving dehalogenation and dehydrogenation, distinct tetrabromoborazine derivatives were produced as reaction intermediates, leading to the formation of specific self-assemblies. Notably, the resulting intricate supramolecular structures include a chiral kagomé lattice composed of molecular dimers exhibiting a unique electronic signature. The final product obtained was a random covalent carbon network with BN-substitution and embedded oxygen heteroatoms. Our study offers valuable insights into the significance of the structure and functionalization of BN precursors in temperature-induced on-surface reactions, which can help future rational precursor design. Additionally, it introduces complex surface architectures that offer a high areal density of borazine cores.
Solution Versus On-Surface Synthesis of Peripherally Oxygen-Annulated Porphyrins through C−O Bond Formation
Joel Deyerling, Dr. Beatrice Berionni Berna, Dr. Dmytro Biloborodov, Dr. Felix Haag, Sena Tömekce, Dr. Marc G. Cuxart, Conghui Li, Prof. Dr. Willi Auwärter, Prof. Dr. Davide Bonifazi
Journal
Angewandte Chemie International Edition
Date
08/2024
Angewandte Chemie International Edition
08/2024
Solution Versus On-Surface Synthesis of Peripherally Oxygen-Annulated Porphyrins through C−O Bond Formation
Joel Deyerling, Dr. Beatrice Berionni Berna, Dr. Dmytro Biloborodov, Dr. Felix Haag, Sena Tömekce, Dr. Marc G. Cuxart, Conghui Li, Prof. Dr. Willi Auwärter, Prof. Dr. Davide Bonifazi
Angewandte Chemie International Edition
28 Aug 2024
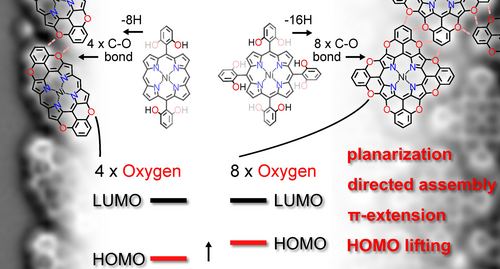
Abstract
This study investigates the synthesis of tetra- and octa-O-fused porphyrinoids employing an oxidative O-annulation approach through C−H activation. Despite encountering challenges such as overoxidation and instability in conventional solution protocols, successful synthesis was achieved on Au(111) surfaces under ultra-high vacuum (UHV) conditions. X-ray photoelectron spectroscopy, scanning tunneling microscopy, and non-contact atomic force microscopy elucidated the preferential formation of pyran moieties via C−O bond formation and subsequent self-assembly driven by C−H⋅⋅⋅O interactions. Furthermore, the O-annulation process was found to reduce the HOMO–LUMO gap by lifting the HOMO energy level, with the effect rising upon increasing the number of embedded O-atoms.
Double Chalcogen Bonding Recognition Arrays in Solution
Dr. Deborah Romito, Prof. Dr. Hanspeter Kählig, Prof. Dr. Paolo Tecilla, Prof. Dr. Gabriele C. Sosso, Prof. Dr. Davide Bonifazi
Journal
Chemistry—A European Journal
Date
07/2024
Chemistry—A European Journal
07/2024
Double Chalcogen Bonding Recognition Arrays in Solution
Dr. Deborah Romito, Prof. Dr. Hanspeter Kählig, Prof. Dr. Paolo Tecilla, Prof. Dr. Gabriele C. Sosso, Prof. Dr. Davide Bonifazi
Chemistry—A European Journal
26 Jul 2024
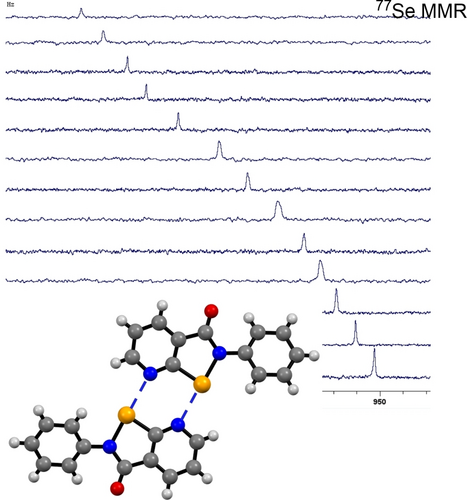
Abstract
N-substituted pyridino-based congeners of Ebselen, named here as Pyrselen, incorporating proximal Se and N atoms, undergo dimerization in solution and the solid state through a dual donor-acceptor arrangement of chalcogen bonding sites. Dimerization constants were measured within the 5–50 M−1 range. Computational studies on the dimers depict a notable charge-transfer contribution to the association, validating Pyrselen as an effective scaffold for designing chalcogen-bonding-based recognition motifs.
One-Step Catalyst-Transfer Macrocyclization: Expanding the Chemical Space of Azaparacyclophanes
Josue Ayuso-Carrillo, Federica Fina, El Czar Galleposo, Ruben R. Ferreira, Pradip Kumar Mondal, Benjamin D. Ward, Davide Bonifazi
DOI: 10.1021/jacs.4c02319
Journal
J. Am. Chem. Soc.
Date
06/2024
J. Am. Chem. Soc.
06/2024
One-Step Catalyst-Transfer Macrocyclization: Expanding the Chemical Space of Azaparacyclophanes
Josue Ayuso-Carrillo, Federica Fina, El Czar Galleposo, Ruben R. Ferreira, Pradip Kumar Mondal, Benjamin D. Ward, Davide Bonifazi
DOI: 10.1021/jacs.4c02319
J. Am. Chem. Soc.
7 Jun 2024
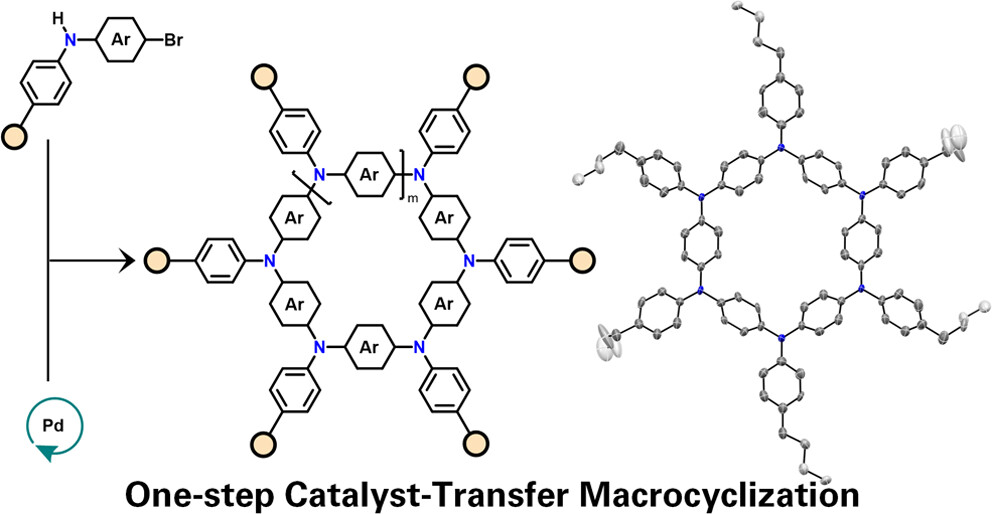
Abstract
In this paper, we report on a one-step catalyst-transfer macrocyclization (CTM) reaction, based on the Pd-catalyzed Buchwald-Hartwig cross-coupling reaction, selectively affording only cyclic structures. This route offers a versatile and efficient approach to synthesize aza[1(n)]paracyclophanes (APCs) featuring diverse functionalities and lumens. The method operates at mild reaction temperatures (40 degrees C) and short reaction times (similar to 2 h), delivering excellent isolated yields (>75% macrocycles) and up to 30% of a 6-membered cyclophane, all under nonhigh-dilution concentrations (35-350 mM). Structural insights into APCs reveal variations in product distribution based on different endocyclic substituents, with steric properties of exocyclic substituents having minimal influence on the macrocyclization. Aryl-type endocyclic substituents predominantly yield 6-membered macrocycles, while polycyclic aromatic units such as fluorene and carbazole favor 4-membered species. Experimental and computational studies support a proposed mechanism of ring-walking catalyst transfer that promotes the macrocycle formation. It has been found that the macrocyclization is driven by the formation of cyclic conformers during the oligomerization step favoring an intramolecular C-N bond formation that, depending on the cycle size, hinges on either preorganization effect or kinetic increase of the reductive elimination step or a combination of the two. The CTM process exhibits a living behavior, facilitating sequential synthesis of other macrocycles by introducing relevant monomers, thus providing a practical synthetic platform for chemical libraries. Notably, CTM operates both under diluted and concentrated regimes, offering scalability potential, unlike typical macrocyclization reactions usually operating in the 0.1-1 mM range.
On-Surface Molecular Recognition Driven by Chalcogen Bonding
Luca Camilli, Conor Hogan, Deborah Romito, Luca Persichetti, Antonio Caporale, Maurizia Palummo, Marco Di Giovannantonio, Davide Bonifazi
Journal
JACS Au
Date
06/2024
JACS Au
06/2024
On-Surface Molecular Recognition Driven by Chalcogen Bonding
Luca Camilli, Conor Hogan, Deborah Romito, Luca Persichetti, Antonio Caporale, Maurizia Palummo, Marco Di Giovannantonio, Davide Bonifazi
JACS Au
5 Jun 2024
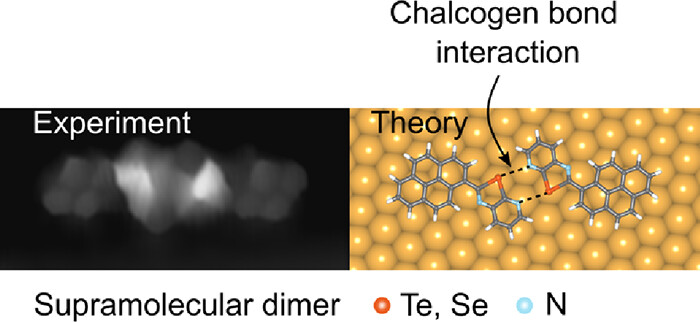
Abstract
Chalcogen bonding interactions (ChBIs) have been widely employed to create ordered noncovalent assemblies in solids and liquids. Yet, their ability to engineer molecular self-assembly on surfaces has not been demonstrated. Here, we report the first demonstration of on-surface molecular recognition solely governed by ChBIs. Scanning tunneling microscopy and ab initio calculations reveal that a pyrenyl derivative can undergo noncovalent chiral dimerization on the Au(111) surface through double ChN interactions involving Te- or Se-containing chalcogenazolo pyridine motifs. In contrast, reference chalcogenazole counterparts lacking the pyridyl moiety fail to form regular self-assemblies on Au, resulting in disordered assemblies.
Celebrating Maurizio Prato’s Passion, Talent and Imagination.
Alberto Bianco, Marcella Bonchio, Davide Bonifazi, Tatiana Da Ros, Michele Maggini, Aurelio Mateo-Alonso, Paolo Tecilla
Journal
Chemistry A European Journal
Date
03/2024
Chemistry A European Journal
03/2024
Celebrating Maurizio Prato’s Passion, Talent and Imagination.
Alberto Bianco, Marcella Bonchio, Davide Bonifazi, Tatiana Da Ros, Michele Maggini, Aurelio Mateo-Alonso, Paolo Tecilla
Chemistry A European Journal
6 Mar 2024

Abstract
You may say I am a dreamer” (Imagine, John Lennon, 1971)
Scientific imagination is a very unique status of mind, the place where new ideas are forged, the very first nucleus of outstanding and surprising discoveries. As such, this is by no means a random idea generator or a casual collection of weakly bonded thoughts. It takes much more to turn a“dreamer” into a visionary scientist. This capacity is rare, as it requires deep commitment, sharp focus, and strong resilience, together with disruptive and contagious creativity, tempered by calm and positive strength, empathy, thoughtfulness, and, above all, empowered by a generous attitude towards opening spaces for collaborative leadership. It takes a whole team to Tango in Science, orchestrated in one cooperative harmony, where every voice of the team members is valued within a full orchestra, tuned to beat at the same rhythm of collaboration. This editorial introduces a Special Collection of papers in honor of Maurizio Prato, showcasing prominent examples of his team‘s work in harmonizing frontier research complexity with groundbreaking achievements in the field of carbon nanostructures (CNS) and molecular nanosciences. The Prato group has established a unique way of playing a broad interdisciplinary research role, tuned at the rhythm of passion and creativity, merging organic and inorganic synthesis with supramolecular chemistry, bioinspired nano-systems, and materials science. Here, the spotlight is on the legacy of Maurizio Prato, listening to the sound of science,being aware of each contribution asolo, but sharing the harmony, strengthening motivation and intensity, making an impact, having fun, and enjoying the melody. Throughout this journey, Maurizio Prato has been considered as one of the most influential scientists for the design and synthesis of tailored CNS as biosustainable functional materials with applications in regenerative medicine and in solar energy research.The Special Collection honors Maurizio Prato with a series of rich contributions highlighting a broad spectrum of research themes that in many ways acknowledge his scientific influence. We encourage the readers to find their ’swing’ in this narrative, taking inspiration for opening new projects and perspectives.
2023
Title
Photoredox Annulation of Polycyclic Aromatic Hydrocarbons
Davide Zanetti, Oliwia Matuszewska, Giuliana Giorgianni, Cristofer Pezzetta, Nicola Demitri, Davide Bonifazi
Journal
JACS Au
Date
11/2023
JACS Au
11/2023
Photoredox Annulation of Polycyclic Aromatic Hydrocarbons
Davide Zanetti, Oliwia Matuszewska, Giuliana Giorgianni, Cristofer Pezzetta, Nicola Demitri, Davide Bonifazi
JACS Au
14 Nov 2023
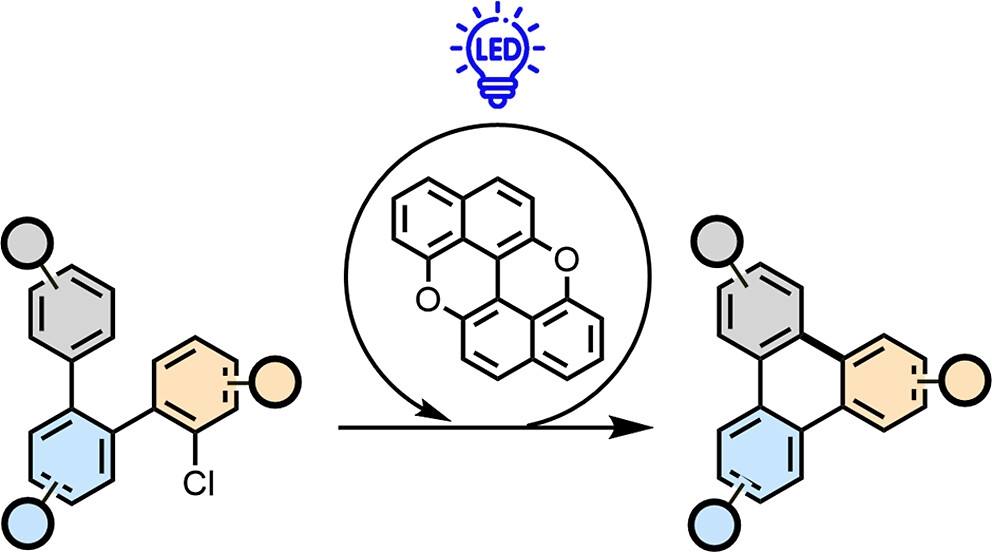
Abstract
The rise of interest in using polycyclic aromatic hydrocarbons (PAHs) and molecular graphenoids in optoelectronics has recently stimulated the growth of modern synthetic methodologies giving access to intramolecular aryl-aryl couplings. Here, we show that a radical-based annulation protocol allows expansion of the planarization approaches to prepare functionalized molecular graphenoids. The enabler of this reaction is peri-xanthenoxanthene, the photocatalyst which undergoes photoinduced single electron transfer with an ortho-oligoarylenyl precursor bearing electron-withdrawing and nucleofuge groups. Dissociative electron transfer enables the formation of persistent aryl radical intermediates, the latter undergoing intramolecular C-C bond formation, allowing the planarization reaction to occur. The reaction conditions are mild and compatible with various electron-withdrawing and -donating substituents on the aryl rings as well as heterocycles and PAHs. The method could be applied to induce double annulation reactions, allowing the synthesis of pi-extended scaffolds with different edge peripheries.
Photoreduction of Anthracenes Catalyzed by peri-Xanthenoxanthene: a Scalable and Sustainable Birch-Type Alternative
Cristian De Luca, Davide Zanetti, Tommaso Battisti, Ruben R. Ferreira, Sofia Lopez, Alexander H. Mcmillan, Sasha Cai Lesher-Perez, Laura Maggini, Davide Bonifazi
Journal
Chem.-Eur. J.
Date
11/2023
Chem.-Eur. J.
11/2023
Photoreduction of Anthracenes Catalyzed by peri-Xanthenoxanthene: a Scalable and Sustainable Birch-Type Alternative
Cristian De Luca, Davide Zanetti, Tommaso Battisti, Ruben R. Ferreira, Sofia Lopez, Alexander H. Mcmillan, Sasha Cai Lesher-Perez, Laura Maggini, Davide Bonifazi
Chem.-Eur. J.
13 Nov 2023
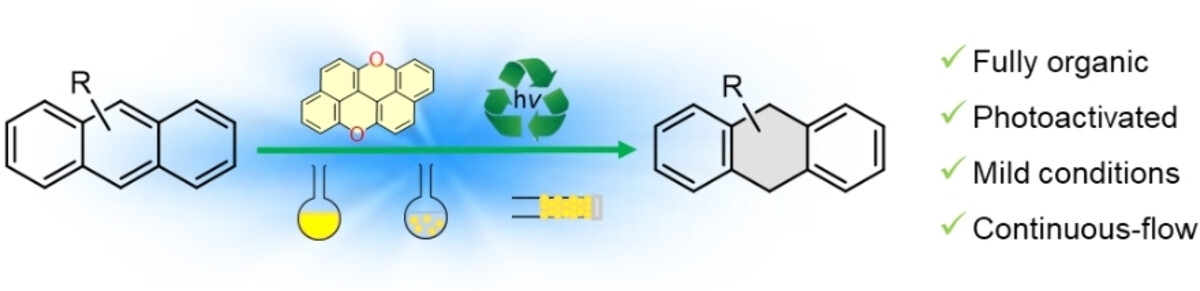
Abstract
The typical Birch reduction transforms arenes into cyclohexa-1,4-dienes by using alkali metals, an alcohol as a proton source, and an amine as solvent. Capitalizing on the strong photoreductive properties of peri-xanthenoxanthene (PXX), herein we report the photocatalyzed Birch-type reduction of acenes by employing visible blue light irradiation at room temperature in the presence of air. Upon excitation at 405 or 460 nm in the presence of a mixture of N,N-diisopropylethylamine (DIPEA) and trifluoromethanesulfonimide (HNTf2) in DMSO, PXX photocatalyzes the selective reduction of full-carbon acene derivatives (24-75 %). Immobilization of PXX onto polydimethylsiloxane (PDMS) beads (PXX-PDMS) allowed the use of the catalyst in heterogeneous batch reactions, giving 9-phenyl-9,10-dihydroanthracene in high yield (68 %). The catalyst could easily be recovered and reused, with no notable drop in performance observed after five reaction cycles. Integration of the PXX-PDMS beads into a microreactor enabled the reduction of acenes under continuous-flow conditions, thereby validating the sustainability and scalability of this heterogeneous-phase approach.
Indacaterol inhibits collective cell migration and IGDQ-mediated single cell migration in metastatic breast cancer MDA-MB-231 cells
Sophie Ayama-Canden, Rodolfo Tondo, Martha Liliana Pineros Leyton, Noelle Ninane, Catherine Demazy, Marc Dieu, Antoine Fattaccioli, Aude Sauvage, Tijani Tabarrant, Stephane Lucas, Davide Bonifazi, Carine Michiels
Journal
Cell Commun. Signal.
Date
10/2023
Cell Commun. Signal.
10/2023
Indacaterol inhibits collective cell migration and IGDQ-mediated single cell migration in metastatic breast cancer MDA-MB-231 cells
Sophie Ayama-Canden, Rodolfo Tondo, Martha Liliana Pineros Leyton, Noelle Ninane, Catherine Demazy, Marc Dieu, Antoine Fattaccioli, Aude Sauvage, Tijani Tabarrant, Stephane Lucas, Davide Bonifazi, Carine Michiels
Cell Commun. Signal.
30 Oct 2023

Abstract
Metastasis is the main cause of deaths related to breast cancer. This is particular the case for triple negative breast cancer. No targeted therapies are reported as efficient until now. The extracellular matrix, in particular the fibronectin type I motif IGDQ, plays a major role in regulating cell migration prior metastasis formation. This motif interacts with specific integrins inducing their activation and the migratory signal transduction.Here, we characterized the migratory phenotype of MDA-MB-231 cells, using functionalized IGDQ-exposing surfaces, and compared it to integrin A5 and integrin B3 knock-down cells. A multiomic analysis was developed that highlighted the splicing factor SRSF6 as a putative master regulator of cell migration and of integrin intracellular trafficking. Indacaterol-induced inhibition of SRSF6 provoked: i) the inhibition of collective and IGDQ-mediated cell migration and ii) ITGA5 sequestration into endosomes and lysosomes. Upon further studies, indacaterol may be a potential therapy to prevent cell migration and reduce metastasis formation in breast cancer.1CRnmBvVXp9LXQy1nJKbUFVideo Abstract
Tweaking the Optoelectronic Properties of S-Doped Polycyclic Aromatic Hydrocarbons by Chemical Oxidation
Oliwia Matuszewska, Tommaso Battisti, Ruben R. Ferreira, Nicolas Biot, Nicola Demitri, Cecile Meziere, Magali Allain, Marc Salle, Samuel Manas-Valero, Eugenio Coronado, Elisa Fresta, Ruben D. Costa, Davide Bonifazi
Journal
Chem.-Eur. J.
Date
02/2023
Chem.-Eur. J.
02/2023
Tweaking the Optoelectronic Properties of S-Doped Polycyclic Aromatic Hydrocarbons by Chemical Oxidation
Oliwia Matuszewska, Tommaso Battisti, Ruben R. Ferreira, Nicolas Biot, Nicola Demitri, Cecile Meziere, Magali Allain, Marc Salle, Samuel Manas-Valero, Eugenio Coronado, Elisa Fresta, Ruben D. Costa, Davide Bonifazi
Chem.-Eur. J.
21 Feb 2023
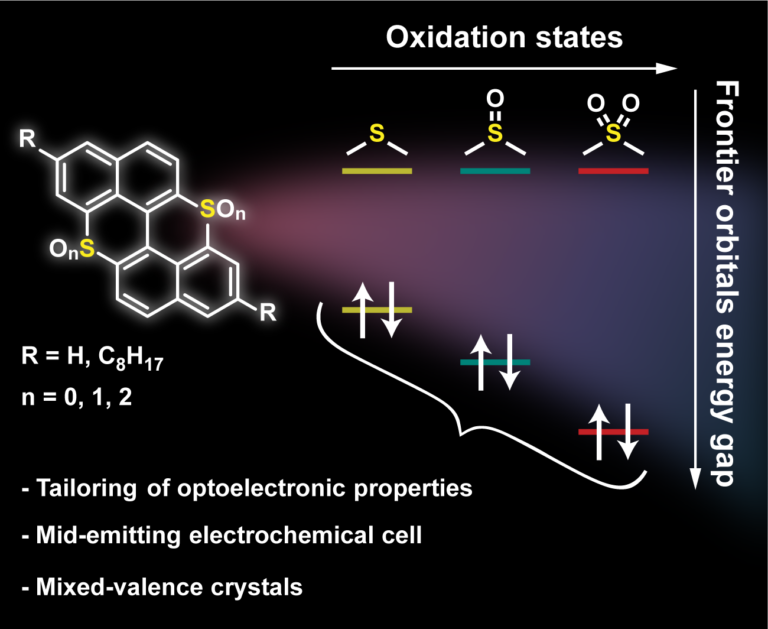
Abstract
Peri-thiaxanthenothiaxanthene, an S-doped analog of peri-xanthenoxanthene, is used as a polycyclic aromatic hydrocarbon (PAH) scaffold to tune the molecular semiconductor properties by editing the oxidation state of the S-atoms. Chemical oxidation of peri-thiaxanthenothiaxanthene with H2O2 led to the relevant sulfoxide and sulfone congeners, whereas electrooxidation gave access to sulfonium-type derivatives forming crystalline mixed valence (MV) complexes. These complexes depicted peculiar molecular and solid-state arrangements with face-to-face pi-pi stacking organization. Photophysical studies showed a widening of the optical bandgap upon progressive oxidation of the S-atoms, with the bis-sulfone derivative displaying the largest value (E-00=2.99 eV). While peri-thiaxanthenothiaxanthene showed reversible oxidation properties, the sulfoxide and sulfone derivatives mainly showed reductive events, corroborating their n-type properties. Electric measurements of single crystals of the MV complexes exhibited a semiconducting behavior with a remarkably high conductivity at room temperature (10(-1)-10(-2) S cm(-1) and 10(-2)-10(-3) S cm(-1) for the O and S derivatives, respectively), one of the highest reported so far. Finally, the electroluminescence properties of the complexes were tested in light-emitting electrochemical cells (LECs), obtaining the first S-doped mid-emitting PAH-based LECs.
Engineering Te-Containing Recognition Modules for Chalcogen Bonding: Towards Supramolecular Polymeric Materials
Deborah Romito, Davide Bonifazi
Journal
Helv. Chim. Acta
Date
02/2023
Helv. Chim. Acta
02/2023
Engineering Te-Containing Recognition Modules for Chalcogen Bonding: Towards Supramolecular Polymeric Materials
1 Feb 2023
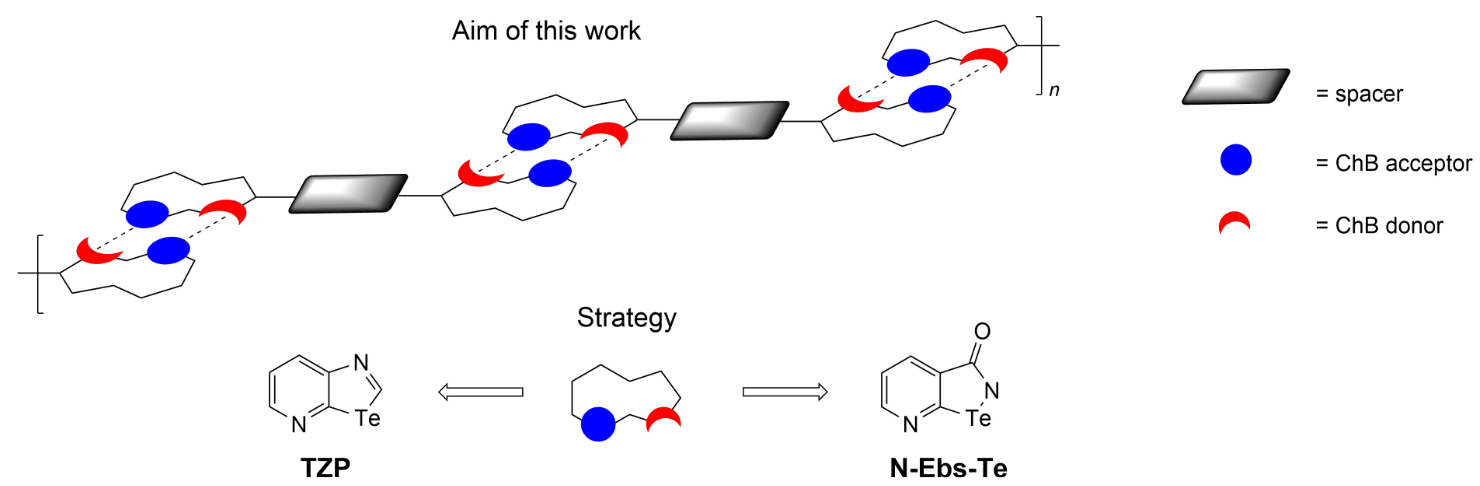
Abstract
Aiming at the preparation of one-dimensional (1D) chalcogen-bonded supramolecular polymers at the solid state, this work describes the different syntheses which have been challenged to obtain ditopic molecular modules. At first, tellurazolopyridyl (TZP) rings have been chosen as recognition units, given their well-proven ability and persistency to self-assemble through double Te center dot center dot center dot N chalcogen bonds (ChBs). The second synthetic strategy dealt with the preparation of pyridyl-modified ebselen Te-containing analogues. By attempting several synthetic protocols, the targeted ebselen derivatives could not be obtained, whereas an unexpected Te-containing lactone as well as a spiro-type Te(IV)-containing derivatives were isolated, with the latter investigated by X-ray diffraction (XRD) analysis.
Expanding the Library of 2-Phenylbenzotellurazoles: Red-Shifting Effect of Ethoxy Functionalities on the UV/Vis Absorption Properties
Deborah Romito, Leonardo Amendolare, Krishnan K. Kalathil, Davide Bonifazi
Journal
Synthesis
Date
01/2023
Synthesis
01/2023
Expanding the Library of 2-Phenylbenzotellurazoles: Red-Shifting Effect of Ethoxy Functionalities on the UV/Vis Absorption Properties
Deborah Romito, Leonardo Amendolare, Krishnan K. Kalathil, Davide Bonifazi
Synthesis
19 Jan 2023
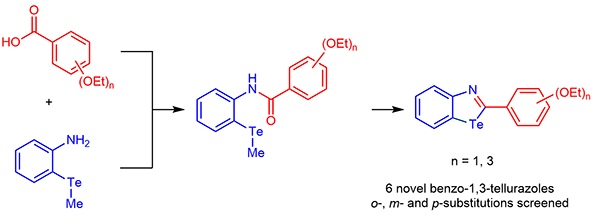
Abstract
This work describes the high-yield synthesis of a novel series of benzotellurazoles bearing a phenyl ring in 2-position, which is differently functionalized with ethoxy chains. Changing the number and the position of these functional groups determines differences in the self-assembly in the solid state, as well as in the photophysical properties of the targeted molecules. As anticipated by theoretical calculations of the HOMO-LUMO gap of each molecule, the presence of ethoxy chains in nand p-positions determines up to 20 nm red-shifts in the absorption peaks, when compared to unsubstituted benzotellurazole. Similarly, more significant changes are observed in the chemical shifts of Te-125 NMR spectra for those derivatives bearing o- and p-ethoxy functionalization.
2022
Title
Boron Nitride-Doped Polyphenylenic Organogels
Jacopo Dosso, Hamid Oubaha, Francesco Fasano, Sorin Melinte, Jean-Francois Gohy, Colan E. Hughes, Kenneth D. M. Harris, Nicola Demitri, Michela Abrami, Mario Grassi, Davide Bonifazi
Journal
Chem. Mat.
Date
12/2022
Chem. Mat.
12/2022
Boron Nitride-Doped Polyphenylenic Organogels
Jacopo Dosso, Hamid Oubaha, Francesco Fasano, Sorin Melinte, Jean-Francois Gohy, Colan E. Hughes, Kenneth D. M. Harris, Nicola Demitri, Michela Abrami, Mario Grassi, Davide Bonifazi
Chem. Mat.
13 Dec 2022
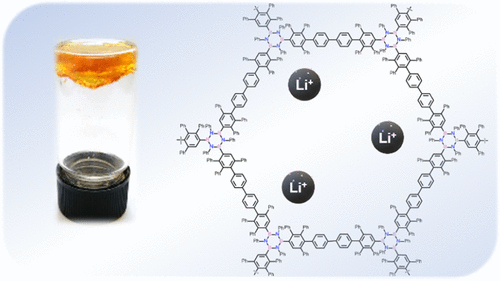
Abstract
Herein, we describe the synthesis of the first boron nitride-doped polyphenylenic material obtained through a [4 + 2] cycloaddition reaction between a triethynyl borazine unit and a biscyclopentadienone derivative, which undergoes organogel formation in chlorinated solvents (the critical jellification concentration is 4% w/w in CHCl3). The polymer has been characterized extensively by Fourier-transform infrared spectroscopy, solid-state C-13 NMR, solid-state B-11 NMR, and by comparison with the isolated monomeric unit. Furthermore, the polymer gels formed in chlorinated solvents have been thoroughly characterized and studied, showing rheological properties comparable to those of polyacrylaniide gels with a low crosslinker percentage. Given the thermal and chemical stability, the material was studied as a potential support for solid-state electrolytes. showing properties comparable to those of polyethylene glycol-based electrolytes, thus presenting great potential for the application of this new class of material in lithium-ion batteries.
HOMO Energy-Level Lifting in p-Type O-Doped Graphenoids: Synthesis of Electrochromic Alkoxy-Decorated Xanthenoxanthenes
Alexandre Rossignon, Beatrice Berionni Berna, A Jorge Parola, César AT Laia, Davide Bonifazi
DOI: 10.1055/a-1976-0291
Journal
Organic Materials
Date
12/2022
Organic Materials
12/2022
HOMO Energy-Level Lifting in p-Type O-Doped Graphenoids: Synthesis of Electrochromic Alkoxy-Decorated Xanthenoxanthenes
Alexandre Rossignon, Beatrice Berionni Berna, A Jorge Parola, César AT Laia, Davide Bonifazi
DOI: 10.1055/a-1976-0291
Organic Materials
12 Dec 2022
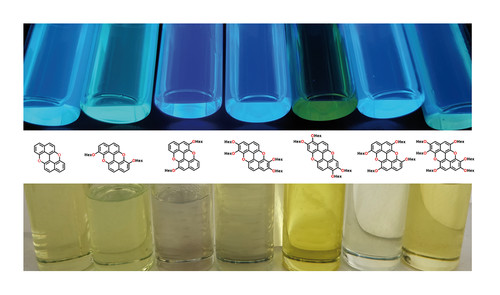
Abstract
A series of novel O-doped polycyclic aromatic hydrocarbons, bearing a different number of electron-donating alkoxy substituents, has been prepared using a novel copper-promoted anaerobic protocol for the cyclisation of highly electron rich peri-xanthenoxanthene molecular modules. The effect of the number and position of the alkoxy substituents on the optoelectronic properties has thus been investigated, unveiling p-type semiconducting properties. All molecules displayed a significant colour change upon oxidation, suggesting that these compounds can be used to devise chromogenic materials to engineer electrochromic devices.
peri-Acenoacene Ribbons with Zigzag BN-Doped Peripheries
Marco Franceschini, Martina Crosta, Ruben R. Ferreira, Daniele Poletto, Nicola Demitri, J. Patrick Zobel, Leticia Gonzalez, Davide Bonifazi
DOI: 10.1021/jacs.2c06803
Journal
J. Am. Chem. Soc.
Date
11/2022
J. Am. Chem. Soc.
11/2022
peri-Acenoacene Ribbons with Zigzag BN-Doped Peripheries
Marco Franceschini, Martina Crosta, Ruben R. Ferreira, Daniele Poletto, Nicola Demitri, J. Patrick Zobel, Leticia Gonzalez, Davide Bonifazi
DOI: 10.1021/jacs.2c06803
J. Am. Chem. Soc.
17 Nov 2022
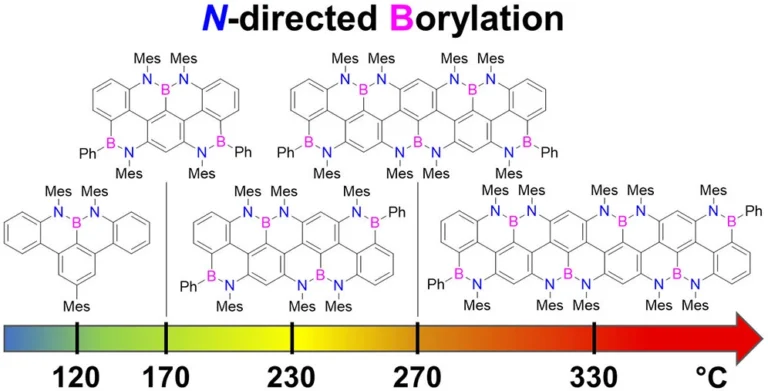
Abstract
Here, we report the synthesis of BN-doped graphenoid nanoribbons, in which peripheral carbon atoms at the zigzag edges have been selectively replaced by boron and nitrogen atoms as BN and NBN motifs. This includes high-yielding ring closure key steps that, through N-directed borylation reaction using solely BBr3, allow the planarization of meta-oligoarylenyl precursors, through the formation of B-N and B-C bonds, to give ter-, quater-, quinque-, and sexi-arylenyl nanoribbons. X-ray single-crystal diffraction studies confirmed the formation of the BN and NBN motifs and the zigzag-edged topology of the regularly doped ribbons. Steady-state absorption and emission investigations at room temperature showed a systematic bathochromic shift of the UV-vis absorption and emission envelopes upon elongation of the oligoarylenyl backbone, with the nanoribbon emission featuring a TADF component. All derivatives displayed phosphorescence at 77 K. Electrochemical studies showed that the pi-extension of the peri-acenoacene framework provokes a lowering of the first oxidative event (from 0.83 to 0.40 V), making these nanoribbons optimal candidates to engineer p-type organic semiconductors.
Supramolecular Chalcogen-Bonded Semiconducting Nanoribbons at Work in Lighting Devices
Deborah Romito, Elisa Fresta, Luca M. Cavinato, Hanspeter Kahlig, Heinz Amenitsch, Laura Caputo, Yusheng Chen, Paolo Samori, Jean-Christophe Charlier, Ruben D. Costa, Davide Bonifazi
Journal
Angew. Chem.-Int. Edit.
Date
09/2022
Angew. Chem.-Int. Edit.
09/2022
Supramolecular Chalcogen-Bonded Semiconducting Nanoribbons at Work in Lighting Devices
Deborah Romito, Elisa Fresta, Luca M. Cavinato, Hanspeter Kahlig, Heinz Amenitsch, Laura Caputo, Yusheng Chen, Paolo Samori, Jean-Christophe Charlier, Ruben D. Costa, Davide Bonifazi
Angew. Chem.-Int. Edit.
19 Sep 2022

Abstract
This work describes the design and synthesis of a pi-conjugated telluro[3,2-beta][1]-tellurophene-based synthon that, embodying pyridyl and haloaryl chalcogen-bonding acceptors, self-assembles into nanoribbons through chalcogen bonds. The ribbons pi-stack in a multi-layered architecture both in single crystals and thin films. Theoretical studies of the electronic states of chalcogen-bonded material showed the presence of a local charge density between Te and N atoms. OTFT-based charge transport measurements showed hole-transport properties for this material. Its integration as a p-type semiconductor in multi-layered Cu-I-based light-emitting electrochemical cells (LECs) led to a 10-fold increase in stability (38 h vs. 3 h) compared to single-layered devices. Finally, using the reference tellurotellurophene congener bearing a C-H group instead of the pyridyl N atom, a herringbone solid-state assembly is formed without charge transport features, resulting in LECs with poor stabilities (<1 h).
IGDQ motogenic peptide gradient induces directional cell migration through integrin ( ?v)?3 activation in MDA-MB-231 metastatic breast cancer cells *
Sophie Ayama-Canden, Rodolfo Tondo, Liliana Pineros, Noelle Ninane, Catherine Demazy, Marc Dieu, Antoine Fattaccioli, Tijani Tabarrant, Stephane Lucas, Davide Bonifazi, Carine Michiels
Journal
Neoplasia
Date
09/2022
Neoplasia
09/2022
IGDQ motogenic peptide gradient induces directional cell migration through integrin ( ?v)?3 activation in MDA-MB-231 metastatic breast cancer cells *
Sophie Ayama-Canden, Rodolfo Tondo, Liliana Pineros, Noelle Ninane, Catherine Demazy, Marc Dieu, Antoine Fattaccioli, Tijani Tabarrant, Stephane Lucas, Davide Bonifazi, Carine Michiels
Neoplasia
1 Sep 2022
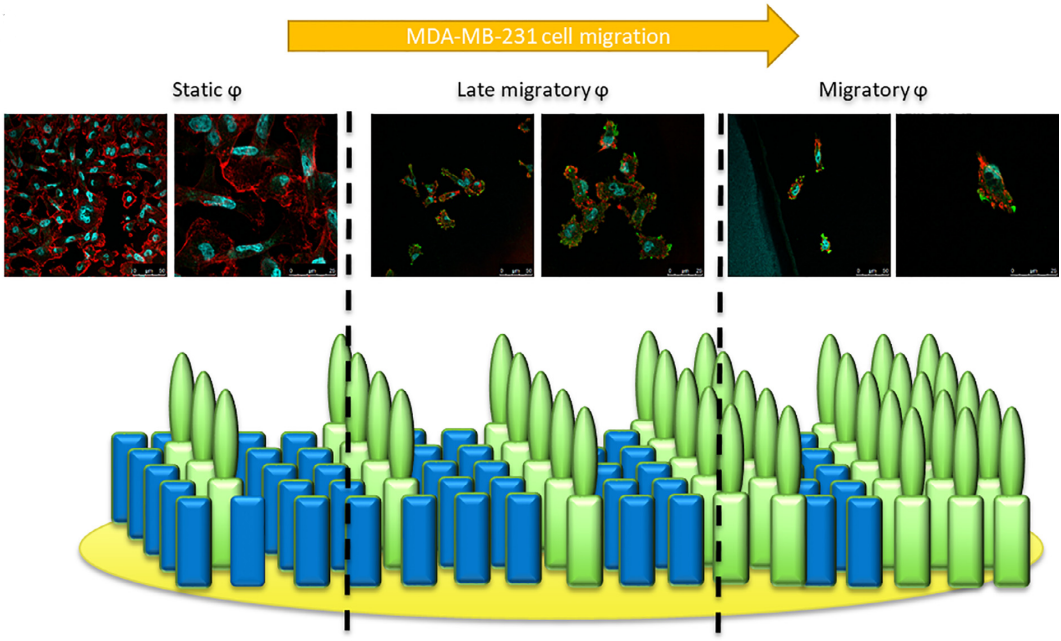
Abstract
In the context of breast cancer metastasis study, we have shown in an in vitro model of cell migration that IGDQ-exposing (IsoLeuGly-Asp-Glutamine type I Fibronectin motif ) monolayers (SAMs) on gold sustain the adhesion of breast cancer MDA-MB-231 cells by triggering Focal Adhesion Kinase and integrin activation. Such tunable scaffolds are used to mimic the tumor extracellular environment, inducing and controlling cell migration. The observed migratory behavior induced by the IGDQ-bearing peptide gradient along the surface allows to separate cell subpopulations with a stationary or migratory phenotype. In this work, we knocked down the integrins alpha 5( beta 1) and ( alpha v) beta since they are already known to be implicated in cell migration. To this aim, a whole proteomic analysis was performed in beta 3 integrin (ITGB3) or alpha 5 integrin (ITGA5) knock-down MDA-MB-231 cells, in order to highlight the pathways implied in the integrin-dependent cell migration. Our results showed that i) ITGB3 depletion influenced ITGA5 mRNA expression, ii) ITGB3 and ITGA5 were both necessary for IGDQ-mediated directional single cell migration and iii) integrin ( alpha v) beta 3 was activated by IGDQ fibronectin type I motif. Finally, the proteomic analysis suggested that co-regulation of recycling transport of ITGB3 by ITGA5 is potentially necessary for directional
Customising excitation properties of polycyclic aromatic hydrocarbons by rational positional heteroatom doping: the peri-xanthenoxanthene (PXX) case
Cataldo Valentini, Duncan Gowland, C. Grazia Bezzu, Deborah Romito, Nicola Demitri, Nicola Bonini, Davide Bonifazi
DOI: 10.1039/d2sc01038k
Journal
Chem. Sci.
Date
06/2022
Chem. Sci.
06/2022
Customising excitation properties of polycyclic aromatic hydrocarbons by rational positional heteroatom doping: the peri-xanthenoxanthene (PXX) case
Cataldo Valentini, Duncan Gowland, C. Grazia Bezzu, Deborah Romito, Nicola Demitri, Nicola Bonini, Davide Bonifazi
DOI: 10.1039/d2sc01038k
Chem. Sci.
1 Jun 2022
Abstract
In this paper we tackle the challenge of gaining control of the photophysical properties of PAHs through a site-specific N-doping within the structural aromatic framework. By developing a simple predictive tool that identifies C(sp(2))-positions that if substituted with a heteroatom would tailor the changes in the absorption and emission spectral envelopes, we predict optimal substitutional patterns for the model peri-xanthenoxanthene (PXX) PAH. Specifically, TDDFT calculations of the electron density difference between the S-1 excited state and S-0 ground state of PXX allowed us to identify the subtleties in the role of sites i .e , electron donating or withdrawing character on excitation. The replacement of two C(sp(2))-atoms with two N-atoms, in either electron donating or withdrawing positions, shifts the electronic transitions either to low or high energy, respectively. This consequently shifts the PXX absorption spectral envelop bathochromically or hypsochromically, as demonstrated by steady-state absorption spectroscopic measurements. Within the series of synthesised N-doped PXX, we tune the optical band gap within an interval of similar to 0.4 eV, in full agreement with the theoretical predictions. Relatedly, measurements show the more blueshifted the absorption/emission energies, the greater the fluorescence quantum yield value (from similar to 45% to similar to 75%). On the other hand, electrochemical investigations suggested that the N-pattern has a limited influence on the redox properties. Lastly, depending on the N-pattern, different supramolecular organisations could be obtained at the solid-state, with the 1,7-pattern PXX molecule forming multi-layered, graphene-like, supramolecular sheets through a combination of weak H-bonding and pi-pi stacking interactions. Supramolecular striped patterned sheets could also be formed with the 3,9- and 4,10-congeners when co-crystallized with a halogen-bond donor molecule.
On-Surface Synthesis of Rigid Benzenoid- and Nonbenzenoid-Coupled Porphyrin-Graphene Nanoribbon Hybrids
Joel Deyerling, Mathias Poertner, Luka Dordevic, Alexander Riss, Davide Bonifazi, Willi Auwaerter
Journal
J. Phys. Chem. C
Date
05/2022
J. Phys. Chem. C
05/2022
On-Surface Synthesis of Rigid Benzenoid- and Nonbenzenoid-Coupled Porphyrin-Graphene Nanoribbon Hybrids
Joel Deyerling, Mathias Poertner, Luka Dordevic, Alexander Riss, Davide Bonifazi, Willi Auwaerter
J. Phys. Chem. C
19 May 2022
Abstract
On-surface synthesis made the fabrication of extended,atomically precise pi-conjugated nanostructures on solid supports possible,with graphene nanoribbons (GNRs) and porphyrin-derived oligomersstanding out. To date, examples combining these two prominent materialclasses are scarce, even though the chemically versatile porphyrins and theatomistic details of the nanographene spacers promise an easy tunability ofstructural and functional properties of the resulting hybrid structures. Here,we report the on-surface synthesis of extended benzenoid- and non-benzenoid-coupled porphyrin-graphene nanoribbon hybrids by sequentialUllmann-type and cyclodehydrogenation reactions of a tailored Zn(II) 5,15-bis(5-bromo-1-naphthyl)porphyrin (Por(BrNaph)2) precursor on Au(111)and Ag(111). Using bond-resolved noncontact atomic force microscopy (nc-AFM) and scanning tunneling microscopy (STM), we characterize thestructures of reaction intermediates and products in detail and provide insight into the effects of the annealing protocol. We furtherdemonstrate the stability and rigidity of the extended one-dimensional porphyrin-GNR oligomers by employing an STM-basedmanipulation procedure, which allows for spectroscopic measurement upon lifting.
Oxygen-Doped PAH Electrochromes: Difurano, Dipyrano, and Furano-Pyrano Containing Naphthalene-Cored Molecules
Jack Fletcher-Charles, Ruben R. Ferreira, Michael Abraham, Deborah Romito, Markus Oppel, Leticia Gonzalez, Davide Bonifazi
Journal
Eur. J. Org. Chem.
Date
01/2022
Eur. J. Org. Chem.
01/2022
Oxygen-Doped PAH Electrochromes: Difurano, Dipyrano, and Furano-Pyrano Containing Naphthalene-Cored Molecules
Jack Fletcher-Charles, Ruben R. Ferreira, Michael Abraham, Deborah Romito, Markus Oppel, Leticia Gonzalez, Davide Bonifazi
Eur. J. Org. Chem.
17 Jan 2022
Abstract
In this work, we report the synthesis of O-doped naphthalene-based electrochromes. Exploiting the CuO-mediated Pummerer oxidative cycloetherification reaction, a series of 1,4- and 1,5-disubstituted naphthalene-cored dipyrano, difurano, and furano-pyrano polycyclic aromatic hydrocarbons (PAHs) have been prepared. Steady-state UV-Vis absorption and emission investigations showed that the spectroscopic profile strongly depends on the O-doping topology, with the dipyrano and the difurano derivatives demonstrating the most red-shifted and blue-shifted electronic transition, respectively. Computational investigations revealed that the cycloetherification reaction raises the HOMO energy level (while the LUMO remains largely unaffected), with the dipyrano derivatives displaying the highest values. Spectroelectrochemical measurements showed that, depending on the O-topology and the type of O-ring, different electrochromic responses could be obtained with colour transitions featuring high contrasts involving yellow, pink, orange or blue colours.
2021
Title
Orbital Mapping of Semiconducting Perylenes on Cu(111)
Giovanni Di Santo, Tanja Miletic, Mathias Schwendt, Yating Zhou, Benson M. Kariuki, Kenneth D. M. Harris, Luca Floreano, Andrea Goldoni, Peter Puschnig, Luca Petaccia, Davide Bonifazi
Journal
J. Phys. Chem. C
Date
11/2021
J. Phys. Chem. C
11/2021
Orbital Mapping of Semiconducting Perylenes on Cu(111)
Giovanni Di Santo, Tanja Miletic, Mathias Schwendt, Yating Zhou, Benson M. Kariuki, Kenneth D. M. Harris, Luca Floreano, Andrea Goldoni, Peter Puschnig, Luca Petaccia, Davide Bonifazi
J. Phys. Chem. C
11 Nov 2021
Abstract
Semiconducting O-doped polycyclic aromatic hydrocarbons constitute a class of molecules whose optoelectronic properties can be tailored by acting on the pi-extension of the carbon-based frameworks and on the oxygen linkages. Although much is known about their photophysical and electrochemical properties in solution, their self-assembly interfacial behavior on solid substrates has remained unexplored so far. In this paper, we have focused our attention on the on-surface self-assembly of O-doped biperylene derivatives. Their ability to assemble in ordered networks on Cu(111) single-crystalline surfaces allowed a combination of structural, morphological, and spectroscopic studies. In particular, the exploitation of the orbital mapping methodology based on angle-resolved photoemission spectroscopy, with the support of scanning tunneling microscopy and low-energy electron diffraction, allowed the identification of both the electronic structure of the adsorbates and their geometric arrangement. Our multi-technique experimental investigation includes the structure determination from powder X-ray diffraction data for a specific compound and demonstrates that the electronic structure of such large molecular self-assembled networks can be studied using the reconstruction methods of molecular orbitals from photoemission data even in the presence of segregated chiral domains.
peri-Xanthenoxanthene (PXX): a Versatile Organic Photocatalyst in Organic Synthesis
Cristofer Pezzetta, Andrea Folli, Oliwia Matuszewska, Damien Murphy, Robert W. M. Davidson, Davide Bonifazi
Journal
Adv. Synth. Catal.
Date
10/2021
Adv. Synth. Catal.
10/2021
peri-Xanthenoxanthene (PXX): a Versatile Organic Photocatalyst in Organic Synthesis
Cristofer Pezzetta, Andrea Folli, Oliwia Matuszewska, Damien Murphy, Robert W. M. Davidson, Davide Bonifazi
Adv. Synth. Catal.
19 Oct 2021
Abstract
Recent years have witnessed a continuous development of photocatalysts to satisfy the growing demand of photophysical and redox properties in photoredox catalysis, with complex structures or alternative strategies devised to access highly reducing or oxidising systems. We report herein the use of peri-xanthenoxanthene (PXX), a simple and inexpensive dye, as an efficient photocatalyst. Its highly reducing excited state allows activation of a wide range of substrates, thus triggering useful radical reactions. Benchmark transformations such as the addition of organic radicals, generated by photoreduction of organic halides, to radical traps are initially demonstrated. More complex dual catalytic manifolds are also shown to be accessible: the beta-arylation of cyclic ketones is successful when using a secondary amine as organocatalyst, while cross-coupling reactions of aryl halides with amines and thiols are obtained when using a Ni co-catalyst. Application to the efficient two-step synthesis of the expensive fluoro-tetrahydro-1H-pyrido[4,3-b]indole, a crucial synthetic intermediate for the investigational drug setipiprant, has been also demonstrated.
Harnessing Selectivity and Sensitivity in Ion Sensing via Supramolecular Recognition: A 3D Hybrid Gold Nanoparticle Network Chemiresistor
Veronica Montes-Garcia, Rafael Furlan de Oliveira, Ye Wang, Andrey Berezin, Pablo Fanjul-Bolado, Maria Begona Gonzalez Garcia, Thomas M. Hermans, Davide Bonifazi, Stefano Casalini, Paolo Samori
Journal
Adv. Funct. Mater.
Date
03/2021
Adv. Funct. Mater.
03/2021
Harnessing Selectivity and Sensitivity in Ion Sensing via Supramolecular Recognition: A 3D Hybrid Gold Nanoparticle Network Chemiresistor
Veronica Montes-Garcia, Rafael Furlan de Oliveira, Ye Wang, Andrey Berezin, Pablo Fanjul-Bolado, Maria Begona Gonzalez Garcia, Thomas M. Hermans, Davide Bonifazi, Stefano Casalini, Paolo Samori
Adv. Funct. Mater.
1 Mar 2021
Abstract
The monitoring of K+ in saliva, blood, urine, or sweat represents a future powerful alternative diagnostic tool to prevent various diseases. However, several K+ sensors are unable to meet the requirements for the development of point-of-care (POC) sensors. To tackle this grand-challenge, the fabrication of chemiresistors (CRs) based on 3D networks of Au nanoparticles covalently bridged by ad-hoc supramolecular receptors for K+, namely dithiomethylene dibenzo-18-crown-6 ether is reported here. A multi-technique characterization allows optimizing a new protocol for fabricating high-performing CRs for real-time monitoring of K+ in complex aqueous environments. The sensor shows exceptional figures of merit: i) linear sensitivity in the 10(-3) to 10(-6) m concentration range; ii) high selectivity to K+ in presence of interfering cations (Na+, Ca2+, and Mg2+); iii) high shelf-life stability (>45 days); iv) reversibility of K+ binding and release; v) successful device integration into microfluidic systems for real-time monitoring; vi) fast response and recovery times (<18 s), and v) K+ detection in artificial saliva. All these characteristics make the supramolecular CRs a potential tool for future applications as POC devices, especially for health monitoring where the determination of K+ in saliva is pivotal for the early diagnosis of diseases.
BN-Doped Metal-Organic Frameworks: Tailoring 2D and 3D Porous Architectures through Molecular Editing of Borazines
Francesco Fasano, Jacopo Dosso, C. Grazia Bezzu, Mariolino Carta, Francois Kerff, Nicola Demitri, Bao-Lian Su, Davide Bonifazi
Journal
Chem.-Eur. J.
Date
02/2021
Chem.-Eur. J.
02/2021
BN-Doped Metal-Organic Frameworks: Tailoring 2D and 3D Porous Architectures through Molecular Editing of Borazines
Francesco Fasano, Jacopo Dosso, C. Grazia Bezzu, Mariolino Carta, Francois Kerff, Nicola Demitri, Bao-Lian Su, Davide Bonifazi
Chem.-Eur. J.
24 Feb 2021
Abstract
Building on the MOF approach to prepare porous materials, herein we report the engineering of porous BN-doped materials using tricarboxylic hexaarylborazine ligands, which are laterally decorated with functional groups at the full-carbon inner shell. Whilst an open porous 3D entangled structure could be obtained from the double interpenetration of two identical metal frameworks derived from the methyl substituted borazine, the chlorine-functionalised linker undergoes formation of a porous layered 2D honeycomb structure, as shown by single-crystal X-ray diffraction analysis. In this architecture, the borazine cores are rotated by 60 degrees in alternating layers, thus generating large rhombohedral channels running perpendicular to the planes of the networks. An analogous unsubstituted full-carbon metal framework was synthesised for comparison. The resulting MOF revealed a crystalline 3D entangled porous structure, composed by three mutually interpenetrating networks, hence denser than those obtained from the borazine linkers. Their microporosity and CO2 uptake were investigated, with the porous 3D BN-MOF entangled structure exhibiting a large apparent BET specific surface area (1091 m(2) g(-1)) and significant CO2 reversible adsorption (3.31 mmol g(-1)) at 1 bar and 273 K.
Organic room-temperature phosphorescence from halogen-bonded organic frameworks: hidden electronic effects in rigidified chromophores
Jiawang Zhou, Ljiljana Stojanovic, Andrey A. Berezin, Tommaso Battisti, Abigail Gill, Benson M. Kariuki, Davide Bonifazi, Rachel Crespo-Otero, Michael R. Wasielewski, Yi-Lin Wu
DOI: 10.1039/d0sc04646a
Journal
Chem. Sci.
Date
01/2021
Chem. Sci.
01/2021
Organic room-temperature phosphorescence from halogen-bonded organic frameworks: hidden electronic effects in rigidified chromophores
Jiawang Zhou, Ljiljana Stojanovic, Andrey A. Berezin, Tommaso Battisti, Abigail Gill, Benson M. Kariuki, Davide Bonifazi, Rachel Crespo-Otero, Michael R. Wasielewski, Yi-Lin Wu
DOI: 10.1039/d0sc04646a
Chem. Sci.
14 Jan 2021
Abstract
Development of purely organic materials displaying room-temperature phosphorescence (RTP) will expand the toolbox of inorganic phosphors for imaging, sensing or display applications. While molecular solids were found to suppress non-radiative energy dissipation and make the RTP process kinetically favourable, such an effect should be enhanced by the presence of multivalent directional non-covalent interactions. Here we report phosphorescence of a series of fast triplet-forming tetraethyl naphthalene-1,4,5,8-tetracarboxylates. Various numbers of bromo substituents were introduced to modulate intermolecular halogen-bonding interactions. Bright RTP with quantum yields up to 20% was observed when the molecule is surrounded by a Br/O halogen-bonded network. Spectroscopic and computational analyses revealed that judicious heavy-atom positioning suppresses non-radiative relaxation and enhances intersystem crossing at the same time. The latter effect was found to be facilitated by the orbital angular momentum change, in addition to the conventional heavy-atom effect. Our results suggest the potential of multivalent noncovalent interactions for excited-state conformation and electronic control.
Substituent-Controlled Tailoring of Chalcogen-Bonded Supramolecular Nanoribbons in the Solid State
Nicolas Biot, Deborah Romito, Davide Bonifazi
Journal
Cryst. Growth Des.
Date
01/2021
Cryst. Growth Des.
01/2021
Substituent-Controlled Tailoring of Chalcogen-Bonded Supramolecular Nanoribbons in the Solid State
6 Jan 2021
Abstract
In this work, we design and synthesize supramolecular 2,5-substituted chalcogenazolo[5,4-beta]pyridine (CGP) synthons arranging in supramolecular ribbons at the solid state. A careful choice of the combination of substituents at the 2- and 5-positions on the CGP scaffold is outlined to accomplish supramolecular materials by means of multiple hybrid interactions, comprising both chalcogen and hydrogen bonds. Depending on the steric and electronic properties of the substituents, different solid-state arrangements have been achieved. Among the different moieties on the 5-position, an oxazole unit has been incorporated on the Se- and Te-congeners by Pd-catalyzed cross-coupling reaction and a supramolecular ribbon-like organization was consistently obtained at the solid state.
2020
Title
1,8,10-Trisubstituted anthracenyl hydrocarbons: Towards versatile scaffolds for multiple-H-bonded recognition arrays
Silvia Forensi, Antoine Stopin, Federica de Leo, Johan Wouters, Davide Bonifazi
Journal
Tetrahedron
Date
12/2020
Tetrahedron
12/2020
1,8,10-Trisubstituted anthracenyl hydrocarbons: Towards versatile scaffolds for multiple-H-bonded recognition arrays
Silvia Forensi, Antoine Stopin, Federica de Leo, Johan Wouters, Davide Bonifazi
Tetrahedron
18 Dec 2020
Abstract
In this work, we describe the synthesis of 1,8,10-trisubstituted anthracenyl scaffolds that, bearing boronic acid functionalities, can act as multiple H-bonding donor systems. The trisubstituted anthracenyl derivatives are synthesized following two main synthetic pathways. Whereas in the first approach trisubstituted anthracenyl derivatives are prepared through the regioselective addition of the relevant organomagnesium nucleophile to 1,8-dichloroanthraquinone, in the second avenue a triflate-bearing anthracene is prepared by reduction of the anthraquinone into the anthrone precursor and functionalized through metal-catalysed cross-coupling reactions. Complementary studies of the Na2S2O4-mediated reduction of 1,8-dichloroanthraquinone allowed to shed further light on the possible mechanism of formation of the anthrone precursor, suggesting the presence of a cis-diol intermediate undergoing antiperiplanar elimination. Solid-state X-ray diffraction investigations of the bisboronic acids show that the molecules self-assemble into dimers through the formation of four H-1-bonds established between the anti-syn conformers of the boronic acid moieties. H-1-NMR titrations between bisboronic acids and tetra H-bond acceptor, diisoquinolino-naphthyridine, showed a significant shift of the -B(OH)(2) proton resonances, suggesting the presence of H-bonding interactions between both molecules. (C) 2020 Published by Elsevier Ltd.
Synthetic strategies tailoring colours in multichromophoric organic nanostructures
Olesia Kulyk, Lou Rocard, Laura Maggini, Davide Bonifazi
DOI: 10.1039/c9cs00555b
Journal
Chem. Soc. Rev.
Date
12/2020
Chem. Soc. Rev.
12/2020
Synthetic strategies tailoring colours in multichromophoric organic nanostructures
7 Dec 2020
Abstract
There has never been a time when colour did not fascinate humanity, inspiring an unceasing manufacturing of a kaleidoscopic variety of dyes and pigments that brought about great revolutions in art, cosmetics, fashion, and our lifestyle as a whole. Over the centuries these tints evolved from raw earths to molecular masterpieces devised by expert chemists whose properties are now being exploited far beyond traditional applications. Mimicking Nature, a timely challenge, regards the preparation of innovative and highly efficient multi-coloured architectures structured at the molecular and nanoscopic scale with specific light-absorbing and light-emitting properties. This tutorial review provides an overview on the chemical strategies developed to engineer and customise these ingenious coloured nanostructures tackling the current performance of organic matter in cutting edge technological sectors, such as solar energy conversion.
Targeting G Protein-Coupled Receptors with Magnetic Carbon Nanotubes: The Case of the A3Adenosine Receptor
Florent Pineux, Stephanie Federico, Karl-Norbert Klotz, Sonja Kachler, Carine Michiels, Mattia Sturlese, Maurizio Prato, Giampiero Spalluto, Stefano Moro, Davide Bonifazi
Journal
ChemMedChem
Date
10/2020
ChemMedChem
10/2020
Targeting G Protein-Coupled Receptors with Magnetic Carbon Nanotubes: The Case of the A3Adenosine Receptor
Florent Pineux, Stephanie Federico, Karl-Norbert Klotz, Sonja Kachler, Carine Michiels, Mattia Sturlese, Maurizio Prato, Giampiero Spalluto, Stefano Moro, Davide Bonifazi
ChemMedChem
19 Oct 2020
Abstract
The A(3)adenosine receptor (AR) is a G protein-coupled receptor (GPCR) overexpressed in the membrane of specific cancer cells. Thus, the development of nanosystems targeting this receptor could be a strategy to both treat and diagnose cancer. Iron-filled carbon nanotubes (CNTs) are an optimal platform for theranostic purposes, and the use of a magnetic field can be exploited for cancer magnetic cell sorting and thermal therapy. In this work, we have conjugated an A(3)AR ligand on the surface of iron-filled CNTs with the aim of targeting cells overexpressing A(3)ARs. In particular, two conjugates bearing PEG linkers of different length were designed. A docking analysis of A(3)AR showed that neither CNT nor linker interferes with ligand binding to the receptor; this was confirmed byin vitropreliminary radioligand competition assays on A(3)AR. Encouraged by this result, magnetic cell sorting was applied to a mixture of cells overexpressing or not the A(3)AR in which our compound displayed indiscriminate binding to all cells. Despite this, it is the first time that a GPCR ligand has been anchored to a magnetic nanosystem, thus it opens the door to new applications for cancer treatment.
Self-assembly and spectroscopic fingerprints of photoactive pyrenyl tectons on hBN/Cu(111)
Domenik M. Zimmermann, Knud Seufert, Luka Dordevic, Tobias Hoh, Sushobhan Joshi, Tomas Marangoni, Davide Bonifazi, Willi Auwaerter
Journal
Beilstein J. Nanotechnol.
Date
09/2020
Beilstein J. Nanotechnol.
09/2020
Self-assembly and spectroscopic fingerprints of photoactive pyrenyl tectons on hBN/Cu(111)
Domenik M. Zimmermann, Knud Seufert, Luka Dordevic, Tobias Hoh, Sushobhan Joshi, Tomas Marangoni, Davide Bonifazi, Willi Auwaerter
Beilstein J. Nanotechnol.
29 Sep 2020
Abstract
The controlled modification of electronic and photophysical properties of polycyclic aromatic hydrocarbons by chemical functionalization, adsorption on solid supports, and supramolecular organization is the key to optimize the application of these compounds in (opto)electronic devices. Here, we present a multimethod study comprehensively characterizing a family of pyridin-4-ylethynyl-functionalized pyrene derivatives in different environments. UV-vis measurements in toluene solutions revealed absorption at wavelengths consistent with density functional theory (DFT) calculations, while emission experiments showed a high fluorescence quantum yield. Scanning tunneling microscopy (STM) and spectroscopy (STS) measurements of the pyrene derivatives adsorbed on a Cu(111)-supported hexagonal boron nitride (hBN) decoupling layer provided access to spatially and energetically resolved molecular electronic states. We demonstrate that the pyrene electronic gap is reduced with an increasing number of substituents. Furthermore, we discuss the influence of template-induced gating and supramolecular organization on the energies of distinct molecular orbitals. The selection of the number and positioning of the pyridyl termini in tetrasubstituted, trans- and cis-like-disubstituted derivatives governed the self-assembly of the pyrenyl core on the nanostructured hBN support, affording dense-packed arrays and intricate porous networks featuring a kagome lattice.
Origin of the Exclusive Ternary Electroluminescent Behavior of BN-Doped Nanographenes in Efficient Single-Component White Light-Emitting Electrochemical Cells
Elisa Fresta, Jacopo Dosso, Juan Cabanillas-Gonzalez, Davide Bonifazi, Ruben D. Costa
Journal
Adv. Funct. Mater.
Date
08/2020
Adv. Funct. Mater.
08/2020
Origin of the Exclusive Ternary Electroluminescent Behavior of BN-Doped Nanographenes in Efficient Single-Component White Light-Emitting Electrochemical Cells
Elisa Fresta, Jacopo Dosso, Juan Cabanillas-Gonzalez, Davide Bonifazi, Ruben D. Costa
Adv. Funct. Mater.
1 Aug 2020
Abstract
White-light-emitting electrochemical cells (WLECs) still represent a significant milestone, since only a few examples with moderate performances have been reported. Particularly, multiemissive white emitters are highly desired, as a paradigm to circumvent phase separation and voltage-dependent emission color issues that are encountered following host:guest and multilayered approaches. Herein, the origin of the exclusive white ternary electroluminescent behavior of BN-doped nanographenes with a B3N3 doping pattern (hexa-perihexabenzoborazinocoronene) is rationalized, leading to one of the most efficient (approximate to 3 cd A(-1)) and stable-over-days single-component and single-layered WLECs. To date, BN-doped nanographenes have featured blue thermally activated delayed fluorescence (TADF). This doping pattern provides, however, white electroluminescence spanning the whole visible range (x/y CIE coordinates of 0.29-31/0.31-38 and average color rendering index (CRI) of 87) through a ternary emission involving fluorescence and thermally activated dual phosphorescence. This temperature-dependent multiemissive mechanism is operative for both photo- and electroluminescence processes and holds over the device lifespan, regardless of the device architecture, active layer composition, and operating conditions. As such, this work represents a new stepping-stone toward designing a new family of multiemissive white emitters based on BN-doped nanographenes that realizes one of the best-performing single-component white-emitting devices compared to the prior-art.
Revealing the Impact of Heat Generation Using Nanographene-Based Light-Emitting Electrochemical Cells
Elisa Fresta, Jacopo Dosso, Juan Cabanillas-Gonzalez, Davide Bonifazi, Ruben D. Costa
Journal
ACS Appl. Mater. Interfaces
Date
06/2020
ACS Appl. Mater. Interfaces
06/2020
Revealing the Impact of Heat Generation Using Nanographene-Based Light-Emitting Electrochemical Cells
Elisa Fresta, Jacopo Dosso, Juan Cabanillas-Gonzalez, Davide Bonifazi, Ruben D. Costa
ACS Appl. Mater. Interfaces
24 Jun 2020
Abstract
Self-heating in light-emitting electrochemical cells (LECs) has been long overlooked, while it has a significant impact on (i) device chromaticity by changing the electroluminescent band shape, (ii) device efficiency because of thermal quenching and exciton dissociation reducing the external quantum efficiency (EQE), and (iii) device stability because of thermal degradation of excitons and eliminate doped species, phase separation, and collapse of the intrinsic emitting zone. Herein, we reveal, for the first time, a direct relationship between self-heating and the early changes in the device chromaticity as well as the magnitude of the error comparing theoretical/experimental EQEs-that is, an overestimation error of ca. 35% at usual pixel working temperatures of around 50 degrees C. This has been realized in LECs using a benchmark nanographene-that is, a substituted hexa-peri-hexabenzocoronene-as an emerging class of emitters with outstanding device performance compared to the prior art of small-molecule LECs-for example, luminances of 345 cd/m(2) and EQEs of 0.35%. As such, this work is a fundamental contribution highlighting how self-heating is a critical limitation toward the optimization and wide use of LECs.
Chalcogen-bond driven molecular recognition at work
Nicolas Biot, Davide Bonifazi
Journal
Coord. Chem. Rev.
Date
06/2020
Coord. Chem. Rev.
06/2020
Chalcogen-bond driven molecular recognition at work
15 Jun 2020
Abstract
Out of the supramolecular toolbox, Secondary Bonding Interactions (SBIs) have attracted in the last decades the attention of the chemical community as novel non-covalent interactions of choice for a large number of chemical systems. Amongst all SBIs, halogen-bonding (XBIs) and chalcogen-bonding (EBIs) interactions are certainly the most important. However, the use of EBIs have received marginal consideration if compared to that of XBIs. By sieving the most significant examples, this review focuses on the theoretical and experimental studies carried out with EBIs in functional systems. In a systematic way the reader is guided through the most recent and representative examples in which chemists have rationally designed molecular modules that, through EBIs, trigger the initiation of chemical reactions, molecular recognition events in solutions and at the solid state to produce self-assembled and self-organised functional materials at different length scales. The study and understanding of the fundamental geometrical and physical parameters ruling EBIs is at its infancy, and it still needs to establish those principles to rationally design and program synthons that, undergoing molecular recognition through EBIs, allow the development of new tailored materials for applications in the field of optoelectronic, sensing, catalysis, and drug discovery. (C) 2020 Published by Elsevier B.V.
O-Annulation to Polycyclic Aromatic Hydrocarbons: A Tale of Optoelectronic Properties from Five- to Seven-Membered Rings
Luka Dordevic, Domenico Milano, Nicola Demitri, Davide Bonifazi
Journal
Org. Lett.
Date
06/2020
Org. Lett.
06/2020
O-Annulation to Polycyclic Aromatic Hydrocarbons: A Tale of Optoelectronic Properties from Five- to Seven-Membered Rings
Luka Dordevic, Domenico Milano, Nicola Demitri, Davide Bonifazi
Org. Lett.
5 Jun 2020
Abstract
We take advantage of the Pummerer oxidative annulation reaction to extend PAHs through the formation of an intramolecular C-O bond with a suitable phenol substituent. Depending on the peripheral topology of the PAH precursor (e.g., pyrene, boron-dipyrromethene, or perylene diimide) five-, six-, and seven-membered O-containing rings could be obtained. The effect of the O-annulations on the optoelectronic properties were studied by various methods with the pyrano-annulated pyrene and BODIPY derivatives depicting quantitative emission quantum yields.
Boron-Nitrogen-Doped Nanographenes: A Synthetic Tale from Borazine Precursors
Jacopo Dosso, Tommaso Battisti, Benjamin D. Ward, Nicola Demitri, Colan E. Hughes, P. Andrew Williams, Kenneth D. M. Harris, Davide Bonifazi
Journal
Chem.-Eur. J.
Date
05/2020
Chem.-Eur. J.
05/2020
Boron-Nitrogen-Doped Nanographenes: A Synthetic Tale from Borazine Precursors
Jacopo Dosso, Tommaso Battisti, Benjamin D. Ward, Nicola Demitri, Colan E. Hughes, P. Andrew Williams, Kenneth D. M. Harris, Davide Bonifazi
Chem.-Eur. J.
20 May 2020
Abstract
In this work, a comprehensive account of the authors synthetic efforts to prepare borazino-doped hexabenzocoronenes by using the Friedel-Crafts-type electrophilic aromatic substitution is reported. Hexafluoro-functionalized aryl borazines, bearing an ortho fluoride leaving group on each of the N- and B-aryl rings, was shown to lead to cascade-type electrophilic aromatic substitution events in the stepwise C-C bond formation, giving higher yields of borazinocoronenes than those obtained with borazine precursors bearing fluoride leaving groups at the ortho positions of the B-aryl substituents. By using this pathway, an unprecedented boroxadizine-doped PAH featuring a gulf-type periphery could be isolated, and its structure proven by single-crystal X-ray diffraction analysis. Mechanistic studies on the stepwise Friedel-Crafts-type cyclization suggest that the mechanism of the planarization reaction proceeds through extension of the pi system. To appraise the doping effect of the boroxadizine unit on the optoelectronic properties of topology-equivalent molecular graphenes, the all-carbon and pyrylium PAH analogues, all featuring a gulf-type periphery, were also prepared. As already shown for the borazino-doped hexabenzocoronene, the replacement of the central benzene ring by its B3N2O congener widens the HOMO-LUMO gap and dramatically enhances the fluorescence quantum yield.
Non-covalent bridging of bithiophenes through chalcogen bonding grips
Deborah Romito, Nicolas Biot, Francesco Babudri, Davide Bonifazi
DOI: 10.1039/c9nj06202e
Journal
New J. Chem.
Date
05/2020
New J. Chem.
05/2020
Non-covalent bridging of bithiophenes through chalcogen bonding grips
Deborah Romito, Nicolas Biot, Francesco Babudri, Davide Bonifazi
DOI: 10.1039/c9nj06202e
New J. Chem.
7 May 2020
Abstract
In this work, chalcogen functionalized dithiophenes, equipped on both extremities with chalcogen-bonding recognition heterocycles, have been prepared following two synthetic pathways. The insertion of the chalcogenazolo[5,4-beta]pyridine allows the control of the organization at the solid state. X-Ray diffraction analysis of the single crystals, showed that the Te-doped derivatives give the most persistant assemblies, with the molecules arranging at solid-state in wire-like polymeric structures through TeMIDLINE HORIZONTAL ELLIPSISN interactions. As expected, the introduction of the Se and Te atoms, dramatically decreases the emission properties, with the Te-bearing congeners being virtually non emissive.
Configurational Selection in Azobenzene-Based Supramolecular Systems Through Dual-Stimuli Processes
Paolo Tecilla, Davide Bonifazi
Journal
ChemistryOpen
Date
05/2020
ChemistryOpen
05/2020
Configurational Selection in Azobenzene-Based Supramolecular Systems Through Dual-Stimuli Processes
1 May 2020
Abstract
Azobenzene is one of the most studied light-controlled molecular switches and it has been incorporated in a large variety of supramolecular systems to control their structural and functional properties. Given the peculiar isomeric distribution at the photoexcited state (PSS), azobenzene derivatives have been used as photoactive framework to build metastable supramolecular systems that are out of the thermodynamic equilibrium. This could be achieved exploiting the peculiar E/Z photoisomerization process that can lead to isomeric ratios that are unreachable in thermal equilibrium conditions. The challenge in the field is to find molecular architectures that, under given external circumstances, lead to a given isomeric ratio in a reversible and predictable manner, ensuring an ultimate control of the configurational distribution and system composition. By reviewing early and recent works in the field, this review aims at describing photoswitchable systems that, containing an azobenzene dye, display a controlled configurational equilibrium by means of a molecular recognition event. Specifically, examples include programmed photoactive molecular architectures binding cations, anions and H-bonded neutral guests. In these systems the non-covalent molecular recognition adds onto the thermal and light stimuli, equipping the supramolecular architecture with an additional external trigger to select the desired configuration composition.
Combining high-resolution scanning tunnelling microscopy and first-principles simulations to identify halogen bonding
James Lawrence, Gabriele C. Sosso, Luka Dordevic, Harry Pinfold, Davide Bonifazi, Giovanni Costantini
Journal
Nat. Commun.
Date
04/2020
Nat. Commun.
04/2020
Combining high-resolution scanning tunnelling microscopy and first-principles simulations to identify halogen bonding
James Lawrence, Gabriele C. Sosso, Luka Dordevic, Harry Pinfold, Davide Bonifazi, Giovanni Costantini
Nat. Commun.
30 Apr 2020
Abstract
Scanning tunnelling microscopy (STM) is commonly used to identify on-surface molecular self-assembled structures. However, its limited ability to reveal only the overall shape of molecules and their relative positions is not always enough to fully solve a supramolecular structure. Here, we analyse the assembly of a brominated polycyclic aromatic molecule on Au(111) and demonstrate that standard STM measurements cannot conclusively establish the nature of the intermolecular interactions. By performing high-resolution STM with a CO-functionalised tip, we clearly identify the location of rings and halogen atoms, determining that halogen bonding governs the assemblies. This is supported by density functional theory calculations that predict a stronger interaction energy for halogen rather than hydrogen bonding and by an electron density topology analysis that identifies characteristic features of halogen bonding. A similar approach should be able to solve many complex 2D supramolecular structures, and we predict its increasing use in molecular nanoscience at surfaces. Scanning tunnelling microscopy (STM) is commonly used to study 2D molecular self-assembly but is not always enough to fully solve a supramolecular structure. Here, the authors combine a high-resolution version of STM with first-principles simulations to precisely identify halogen bonding in polycyclic aromatic molecules.
Probing Peripheral H-Bonding Functionalities in BN-Doped Polycyclic Aromatic Hydrocarbons
Jonathan Tasseroul, Maria Mercedes Lorenzo-Garcia, Jacopo Dosso, Francois Simon, Simone Velari, Alessandro De Vita, Paolo Tecilla, Davide Bonifazi
Journal
J. Org. Chem.
Date
03/2020
J. Org. Chem.
03/2020
Probing Peripheral H-Bonding Functionalities in BN-Doped Polycyclic Aromatic Hydrocarbons
Jonathan Tasseroul, Maria Mercedes Lorenzo-Garcia, Jacopo Dosso, Francois Simon, Simone Velari, Alessandro De Vita, Paolo Tecilla, Davide Bonifazi
J. Org. Chem.
6 Mar 2020
Abstract
The replacement of carbon atoms at the zigzag periphery of a benzo[fg]tetracenyl derivative with an NBN atomic triad allows the formation of heteroatom-doped polycyclic aromatic hydrocarbon (PAH) isosteres, which expose BN mimics of the amidic NH functions. Their ability to form H-bonded complexes has never been touched so far. Herein, we report the first solution recognition studies of peripherally NBN-doped PAHs to form H-bonded DD center dot AA- and ADDA center dot DAAD-type complexes with suitable complementary H-bonding acceptor partners. The first determination of K-a in solution showed that the 1:1 association strength is around 27 +/- 1 M-1 for the DD center dot AA complexes in C6D6, whereas it rises to 1820 +/- 130 M-1 for the ADDA center dot DAAD array in CDCl3. Given the interest of BN-doped polyaromatic hydrocarbons in supramolecular and materials chemistry, it is expected that these findings will open new possibilities to design novel materials, where the H-bonding properties of peripheral NH hydrogens could serve as anchors to tailor the organizational properties of PAHs.
Concurring Chalcogen- and Halogen-Bonding Interactions in Supramolecular Polymers for Crystal Engineering Applications
Nicolas Biot, Davide Bonifazi
Journal
Chem.-Eur. J.
Date
03/2020
Chem.-Eur. J.
03/2020
Concurring Chalcogen- and Halogen-Bonding Interactions in Supramolecular Polymers for Crystal Engineering Applications
2 Mar 2020
Abstract
The engineering of crystalline molecular solids through the simultaneous combination of distinctive non-covalent interactions is an important field of research, as it could allow chemist to prepare materials depicting multi-responsive properties. It is in this context that, pushed by a will to expand the chemical space of chalcogen-bonding interactions, a concept is put forward for which chalcogen- and halogen-bonding interactions can be used simultaneously to engineer multicomponent co-crystals. Through the rational design of crystallizable molecules, chalcogenazolo pyridine scaffold (CGP) modules were prepared that, bearing either a halogen-bond acceptor or donor at the 2-position, can interact with suitable complementary molecular modules undergoing formation of supramolecular polymers at the solid state. The recognition reliability of the CGP moiety to form chalcogen-bonded dimers allows the formation of heteromolecular supramolecular polymers through halogen-bonding interactions, as confirmed by single-crystal X-ray diffraction analysis.
O-Doped Nanographenes: The Pyrano/Pyrylium Route Towards Semiconducting Cationic Mixed-Valence Complexes
Luka Dordevic, Cataldo Valentini, Nicola Demitri, Cecile Meziere, Magali Allain, Marc Salle, Andrea Folli, Damien Murphy, Samuel Manas-Valero, Eugenio Coronado, Davide Bonifazi
Journal
Angew. Chem.-Int. Edit.
Date
03/2020
Angew. Chem.-Int. Edit.
03/2020
O-Doped Nanographenes: The Pyrano/Pyrylium Route Towards Semiconducting Cationic Mixed-Valence Complexes
Luka Dordevic, Cataldo Valentini, Nicola Demitri, Cecile Meziere, Magali Allain, Marc Salle, Andrea Folli, Damien Murphy, Samuel Manas-Valero, Eugenio Coronado, Davide Bonifazi
Angew. Chem.-Int. Edit.
2 Mar 2020
Abstract
Herein we report an efficient synthesis to prepare O-doped nanographenes derived from the pi-extension of pyrene. The derivatives are highly fluorescent and feature low oxidation potentials. Using electrooxidation, crystals of cationic mixed-valence (MV) complexes were grown in which the organic salts organize into face-to-face pi-stacks, a favorable solid-state arrangement for organic electronics. Variable-temperature electron paramagnetic resonance (EPR) measurements and relaxation studies suggest a strong electron delocalization along the longitudinal axis of the columnar pi-stacking architectures. Electric measurements of single crystals of the MV salts show a semiconducting behavior with a remarkably high conductivity at room temperature. These findings support the notion that pi-extension of heteroatom-doped polycyclic aromatic hydrocarbons is an attractive approach to fabricate nanographenes with a broad spectrum of semiconducting properties and high charge mobilities.
2019
Title
Patterning Porous Networks through Self-Assembly of Programmed Biomacromolecules
Laure-Elie Carloni, C. Grazia Bezzu, Davide Bonifazi
Journal
Chem.-Eur. J.
Date
12/2019
Chem.-Eur. J.
12/2019
Patterning Porous Networks through Self-Assembly of Programmed Biomacromolecules
18 Dec 2019
Abstract
Two-dimensional (2D) porous networks are of great interest for the fabrication of complex organized functional materials for potential applications in nanotechnologies and nanoelectronics. This review aims at providing an overview of bottom-up approaches towards the engineering of 2D porous networks by using biomacromolecules, with a particular focus on nucleic acids and proteins. The first part illustrates how the advancements in DNA nanotechnology allowed for the attainment of complex ordered porous two-dimensional DNA nanostructures, thanks to a biomimetic approach based on DNA molecules self-assembly through specific hydrogen-bond base pairing. The second part focuses the attention on how polypeptides and proteins structural properties could be used to engineer organized networks templating the formation of multifunctional materials. The structural organization of all examples is discussed as revealed by scanning probe microscopy or transmission electron microscopy imaging techniques.
Synthesis of 3,5-Disubstituted Isoxazoles through a 1,3-Dipolar Cycloaddition Reaction between Alkynes and Nitrile Oxides Generated from O-Silylated Hydroxamic Acids
Laure-Elie Carloni, Stefan Mohnani, Davide Bonifazi
Journal
Eur. J. Org. Chem.
Date
11/2019
Eur. J. Org. Chem.
11/2019
Synthesis of 3,5-Disubstituted Isoxazoles through a 1,3-Dipolar Cycloaddition Reaction between Alkynes and Nitrile Oxides Generated from O-Silylated Hydroxamic Acids
30 Nov 2019
Abstract
In this paper, we report the regioselective synthesis of 3,5-disubstituted isoxazoles by 1,3-dipolar cycloaddition between alkynyl dipolarophiles and nitrile oxide dipoles generated in-situ from O-silylated hydroxamic acids in the presence of trifluoromethanesulfonic anhydride and NEt3. Thanks to the mild, metal-free and oxidant-free conditions that this strategy offers, the reaction was successfully applied to a wide variety of alkynyl dipolarophiles, demonstrating the tolerance of this approach to diverse functional groups. In particular, we have shown that the method was compatible with biological molecules such as peptides and peptide nucleic acids (PNA). This protocol constitutes another example of metal-free 1,3-dipolar cycloaddition leading to the regioselective formation of isoxazoles.
Enantioselective Synthesis of N-Benzylic Heterocycles: A Nickel and Photoredox Dual Catalysis Approach
Cristofer Pezzetta, Davide Bonifazi, Robert W. M. Davidson
Journal
Org. Lett.
Date
11/2019
Org. Lett.
11/2019
Enantioselective Synthesis of N-Benzylic Heterocycles: A Nickel and Photoredox Dual Catalysis Approach
Cristofer Pezzetta, Davide Bonifazi, Robert W. M. Davidson
Org. Lett.
15 Nov 2019
Abstract
Reported herein is a dual nickel- and photoredox-catalyzed modular approach for the preparation of enantioenriched N-benzylic heterocycles. alpha-Heterocyclic carboxylic acids, easily obtainable from common commercial material, are reported as suitable substrates for a decarboxylative strategy in conjunction with a chiral pyridine-oxazoline (PyOx) ligand, providing quick access to enantioenriched drug-like products. The presence of a directing group on the heterocyclic moiety is shown to be beneficial, affording improved stereoselectivity in a number of cases.
Kinked Silicon Nanowires: Superstructures by Metal-Assisted Chemical Etching
Georgiana Sandu, Jonathan Avila Osses, Marine Luciano, Darwin Caina, Antoine Stopin, Davide Bonifazi, Jean-Francois Gohy, Alejandro Silhanek, Ileana Florea, Mounib Bahri, Ovidiu Ersen, Philippe Leclere, Sylvain Gabriele, Alexandru Vlad, Sorin Melinte
Journal
Nano Lett.
Date
11/2019
Nano Lett.
11/2019
Kinked Silicon Nanowires: Superstructures by Metal-Assisted Chemical Etching
Georgiana Sandu, Jonathan Avila Osses, Marine Luciano, Darwin Caina, Antoine Stopin, Davide Bonifazi, Jean-Francois Gohy, Alejandro Silhanek, Ileana Florea, Mounib Bahri, Ovidiu Ersen, Philippe Leclere, Sylvain Gabriele, Alexandru Vlad, Sorin Melinte
Nano Lett.
1 Nov 2019
Abstract
We report on metal-assisted chemical etching of Si for the synthesis of mechanically stable, hybrid crystallographic orientation Si superstructures with high aspect ratio, above 200. This method sustains high etching rates and facilitates reproducible results. The protocol enables the control of the number, angle, and location of the kinks via successive etch-quench sequences. We analyzed relevant Au mask catalyst features to systematically assess their impact on a wide spectrum of etched morphologies that can be easily attained and customized by fine-tuning of the critical etching parameters. For instance, the designed kinked Si nanowires can be incorporated in biological cells without affecting their viability. An accessible numerical model is provided to explain the etch profiles and the physicochemical events at the Si/Au-electrolyte interface and offers guidelines for the development of finite-element modeling of metal-assisted Si chemical etching.
O-Annulation Leading to Five-, Six-, and Seven-Membered Cyclic Diaryl Ethers Involving C-H Cleavage
Alexandre Rossignon, Davide Bonifazi
Journal
Synthesis
Date
10/2019
Synthesis
10/2019
O-Annulation Leading to Five-, Six-, and Seven-Membered Cyclic Diaryl Ethers Involving C-H Cleavage
1 Oct 2019
Abstract
Cyclic diaryl ethers are present in multiple natural compounds, organic pollutants as well as in pi-conjugated organic molecular materials. This short review aims at overviewing the main synthetic advances in the O-annulation methods for preparing five-, six-, and seven-membered rings through C-H cleavage. 1 Introduction 2 Five-Membered Rings: The Dibenzofuran (DBF) Motif 2.1 Palladium-Catalysed C-H Activation 2.2 Copper-Catalysed C-H Activation 2.3 Non-CH Activation Oxidant-Mediated Cyclisation 2.4 Light-Mediated Cyclisation 2.5 Acid-catalysed C-O Cleavage/C-O Formation 3 Six-Membered Rings: DBX, PXX, Xanthone, and Their Derivatives 3.1 Dibenzoxanthene (DBX) 3.2 Peri -Xanthenoxanthene (PXX) 3.3 Xanthones 3.4 Miscellaneous 4 Seven-Membered Rings: Cularine 5 Conclusion
Templating Porphyrin Anisotropy via Magnetically Aligned Carbon Nanotubes
Luka Dordevic, Tomas Marangoni, Mingjie Liu, Rita De Zorzi, Silvano Geremia, Andrea Minoia, Roberto Lazzaroni, Yasuhiro Ishida, Davide Bonifazi
Journal
ChemPlusChem
Date
09/2019
ChemPlusChem
09/2019
Templating Porphyrin Anisotropy via Magnetically Aligned Carbon Nanotubes
Luka Dordevic, Tomas Marangoni, Mingjie Liu, Rita De Zorzi, Silvano Geremia, Andrea Minoia, Roberto Lazzaroni, Yasuhiro Ishida, Davide Bonifazi
ChemPlusChem
1 Sep 2019
Abstract
The preparation and characterisation of a novel three-dimensional organic material consisting of porphyrin arrays on carbon nanotubes embedded in an organogel is reported. Firstly, the porphyrin array was prepared through metal-ligand coordination of a ditopic ligand (1,2-bis(4-pyridyl)ethane) and two bis-Zn(II) porphyrins, linked through a pyrene core, and was studied through UV-Vis, NMR and diffusion spectroscopies. Secondly, the porphyrin supramolecular architecture was adsorbed on pristine carbon nanotubes, greatly improving the dispersibility of the latter in organic solvents. The hybrid material was characterised by means of UV-Vis spectroscopy, microscopic techniques and thermogravimetric analysis. Finally, by exploiting the anisotropic magnetic susceptibility of carbon nanotubes, the hybrid material was aligned under a magnetic field, the organisation of which could be maintained by in situ gelation. The resultant hybrid organogel exhibited notable optical anisotropy, suggesting an anisotropic arrangement of the porphyrin-CNTs architectures in the macroscopic material.
Photoactive Boron-Nitrogen-Carbon Hybrids: From Azo-borazines to Polymeric Materials
Hamid Oubaha, Nicola Demitri, Joelle Rault-Berthelot, Philippe Dubois, Olivier Coulembier, Davide Bonifazi
Journal
J. Org. Chem.
Date
07/2019
J. Org. Chem.
07/2019
Photoactive Boron-Nitrogen-Carbon Hybrids: From Azo-borazines to Polymeric Materials
Hamid Oubaha, Nicola Demitri, Joelle Rault-Berthelot, Philippe Dubois, Olivier Coulembier, Davide Bonifazi
J. Org. Chem.
19 Jul 2019
Abstract
In this paper, we describe synthetic routes for preparing a novel switchable BNC-based chromophore, composed of a borazine core peripherally functionalized with azobenzene moieties. Capitalizing on the Pd-catalyzed Suzuki cross-coupling reaction between a tris-triflate borazine and an organoboron azobenzene derivative, a photoswtichable azo-borazine derivative was successfully prepared. The molecule showed reversible E/Z photoisomerization upon irradiation at the maximum of the intense pi-pi* absorption feature (360 nm). X-ray crystallographic investigations revealed a nonplanar orientation of the three azobenzene moieties and the trans configuration of the -N=N- bonds. Building on the synthetic versatility of the borazine-azobenzene derivative, we used this photoactive scaffold to engineer soluble BN-doped polythiophene polymers. Photophysical characterization performed in solvents of different polarity suggested that the polymer undergoes intramolecular charge transfer (ICT).
Structural Properties of Highly Doped Borazino Polyphenylenes Obtained through Condensation Reaction
Jacopo Dosso, Davide Marinelli, Nicola Demitri, Davide Bonifazi
Journal
ACS Omega
Date
05/2019
ACS Omega
05/2019
Structural Properties of Highly Doped Borazino Polyphenylenes Obtained through Condensation Reaction
Jacopo Dosso, Davide Marinelli, Nicola Demitri, Davide Bonifazi
ACS Omega
1 May 2019
Abstract
Here we describe the synthesis and spectroscopic and structural characterization of various borazine-doped polyphenylenes displaying high doping dosages (16-18%). Capitalizing on the condensation reaction approach, the desired products were formed using a mixture of p-phenylendiamine and aniline with BCl3, followed by the addition of an aryl lithium derivative. The use of mesityl lithium (MesLi) yields strained multiborazine derivatives, which proved to be unstable in the presence of moisture. However, when xylyl lithium (XylLi) was used, chemically stable multiborazines were obtained, with oligomers showing molecular weight up to 10(4), corresponding to 16-18 monomer units. While the dimer, trimer, and tetramer could be isolated as pure products and their structure characterized by mass and NMR analysis, higher oligomers could only be isolated as mixtures of B-hydroxy-substituted derivatives and characterized by gel permeation chromatography. The structures of the dimer and trimer derivatives were confirmed by X-ray analysis, which nicely showed the presence of the two and three borazine rings spaced by one and two 1,4-aryl bridges, respectively. Notably, the trimer forms a porous crystalline clathrate. The peripheral xylyl and phenyl moieties of each molecule intramolecularly embrace each other through C-H and p-p stacking interactions. Steady-state UV-vis absorption characterization suggested that the molecules are UV absorbers, with the extinction coefficient linearly scaling with the degree of oligomerization. On the other hand, low-emission quantum yields were obtained for all derivatives (<7%), suggesting that high BN-doping dosages dramatically affect the emission properties of the doped polyphenylenes.
Coverage-Controlled Polymorphism of H-Bonded Networks on Au(111)
Nico Schmidt, Mihaela Enache, Laura Maggini, Remco W. A. Havenith, Davide Bonifazi, Meike Stohr
Journal
J. Phys. Chem. C
Date
03/2019
J. Phys. Chem. C
03/2019
Coverage-Controlled Polymorphism of H-Bonded Networks on Au(111)
Nico Schmidt, Mihaela Enache, Laura Maggini, Remco W. A. Havenith, Davide Bonifazi, Meike Stohr
J. Phys. Chem. C
28 Mar 2019
Abstract
We report on the self-assembly of a conformational flexible organic compound on Au(111) using scanning tunneling microscopy and low-energy electron diffraction measurements. We observed different conformers of the compound upon adsorption on the reconstructed Au(111) surface. Increasing the molecular coverage enhanced the lateral pressure, that is, parallel to the surface, favoring a coverage-controlled transition from a supramolecular network displaying only one molecular organization, into a polymorphic array with two coexisting arrangements. Our results give insights into the role of substrate-induced conformational changes on the formation of polymorphic supramolecular networks.
Leveraging Fluorescent Emission to Unitary Yield: Dimerization of Polycyclic Aromatic Hydrocarbons
Tanja Miletic, Nicolas Biot, Nicola Demitri, Giuseppe Brancato, Benson M. Kariuki, Davide Bonifazi
Journal
Helv. Chim. Acta
Date
03/2019
Helv. Chim. Acta
03/2019
Leveraging Fluorescent Emission to Unitary Yield: Dimerization of Polycyclic Aromatic Hydrocarbons
Tanja Miletic, Nicolas Biot, Nicola Demitri, Giuseppe Brancato, Benson M. Kariuki, Davide Bonifazi
Helv. Chim. Acta
1 Mar 2019
Abstract
We report on the synthesis and characterization of novel substituted 1,1-biperylene-2,2-diols in which the dihedral angle between the two polycyclic aromatic hydrocarbon (PAH) units is tailored from ca. 60 degrees to ca. 90 degrees in the solid state by introduction of cyclo-etheric straps or sterically hindered groups such as the triisopropylsilyl (TIPS) group. Depending on the type of substitution, we lock the dihedral angle between the perylenyl moieties enabling fine-tuning of the molecular optoelectronic properties, with the molecules displaying the smallest angles acting as exceptionally strong emitters with unitary quantum yields.
2018
Title
A benzoxazine/substituted borazine composite coating: A new resin for improving the corrosion resistance of the pristine benzoxazine coating applied on aluminum
Alexis Renaud, Leila Bonnaud, Ludovic Dumas, Tao Zhang, Yoann Paint, Francesco Fasano, Olesia Kulyk, Eva Pospisilova, Bernard Nysten, Arnaud Delcorte, Davide Bonifazi, Philippe Dubois, Marie-Georges Olivier, Marc Poorteman
Journal
Eur. Polym. J.
Date
12/2018
Eur. Polym. J.
12/2018
A benzoxazine/substituted borazine composite coating: A new resin for improving the corrosion resistance of the pristine benzoxazine coating applied on aluminum
Alexis Renaud, Leila Bonnaud, Ludovic Dumas, Tao Zhang, Yoann Paint, Francesco Fasano, Olesia Kulyk, Eva Pospisilova, Bernard Nysten, Arnaud Delcorte, Davide Bonifazi, Philippe Dubois, Marie-Georges Olivier, Marc Poorteman
Eur. Polym. J.
1 Dec 2018
Abstract
In this paper, laboratory synthesized Phenol-paraPhenyleneDiAmine (P-pPDA) benzoxazine containing different amounts of B-trimesityl-N-triphenylborazine was applied by spin coating on aluminum and thermally cured. The addition of the borazine derivative (borazine 1) does not appear to modify the curing characteristics of the P-pPDA matrix itself as shown by FTIR, DSC and DEA analyses; however, some interactions - chemical and/or physical (co-crystallization) - between P-pPDA and borazine 1 cannot be excluded. The microstructure of the composites is characterized by a two phase system consisting of a dispersion of nanosized (10-20 nm) clusters for the lowest borazine 1 concentration (0.5 wt%), evolving towards bigger (100-200 nm), agglomerated clusters for higher borazine 1 concentrations (3 wt%) and finally, continuous, dendritic structures within the P-pPDA matrix for the highest borazine 1 concentration (10 wt%). The benzoxazine composite coating containing 0.5 wt % trimesitylborazine derivative showed a largely increased and durable ability to protect the aluminum substrate. It is shown that a highly capacitive behavior and durable barrier properties can be obtained for P-pPDA coatings containing such a low amount of borazine derivative homogeneously dispersed in the benzoxazine matrix. For concentrations of 3 wt%, as agglomeration took place and dendrites appeared for the highest concentration of borazine derivative (10 wt%), the corrosion resistance decreased with time.
Tailored Synthesis of N-Substituted peri-Xanthenoxanthene Diimide (PXXDI) and Monoimide (PXXMI) Scaffolds
Andrea Sciutto, Andrey Berezin, Matteo Lo Cicero, Tanja Miletic, Antoine Stopin, Davide Bonifazi
Journal
J. Org. Chem.
Date
11/2018
J. Org. Chem.
11/2018
Tailored Synthesis of N-Substituted peri-Xanthenoxanthene Diimide (PXXDI) and Monoimide (PXXMI) Scaffolds
Andrea Sciutto, Andrey Berezin, Matteo Lo Cicero, Tanja Miletic, Antoine Stopin, Davide Bonifazi
J. Org. Chem.
16 Nov 2018
Abstract
The tailored synthesis of homo (A(2)) and hetero (AB) N-substituted peri-xanthenoxanthene diimides (PXXDIs) and peri-functionalized PXX monoimides (PXXMIs) from 3-hydroxy naphthalic anhydride is described. As A(2)-type PXXDIs could be synthesized in one step, AB-type PXXDIs and PXXMIs were prepared through a modular approach capitalizing on sequential Suzuki coupling, imidation, and Pummerer reactions with very high yields. In view of their potential applications as organic semiconductors, self-organization studies were performed through liquid deposition on surfaces, depicting the formation of islands, needles, and rods.
Templated Chromophore Assembly on Peptide Scaffolds: A Structural Evolution
Lou Rocard, Darren Wragg, Samuel Alexander Jobbins, Lorenzo Luciani, Johan Wouters, Stefano Leoni, Davide Bonifazi
Journal
Chem.-Eur. J.
Date
10/2018
Chem.-Eur. J.
10/2018
Templated Chromophore Assembly on Peptide Scaffolds: A Structural Evolution
Lou Rocard, Darren Wragg, Samuel Alexander Jobbins, Lorenzo Luciani, Johan Wouters, Stefano Leoni, Davide Bonifazi
Chem.-Eur. J.
26 Oct 2018
Abstract
The use of a template that bears pre-programmed receptor sites for selectively accommodating chromophores at given positions is an attractive approach for engineering artificial-light-harvesting systems. Indulging this line of thought, this work tackles the creation of tailored antenna architectures with yellow, red and blue chromophores, exploiting three dynamic covalent reactions simultaneously, namely disulfide exchange, acyl hydrazone, and boronic ester formations. The effect of various structural modifications, such as the chromophores as well as their spatial organization (distance, orientation, order) on the energy transfer within the antennas was studied by means of steady-state UV/Vis absorption and fluorescence spectroscopies. This systematic study allowed for a significant improvement of the energy-transfer efficiencies to a noticeable 22 and 15% for the yellow and red donors, respectively, across the chromophores to the blue acceptor. Metadynamics simulations suggested that the conformational properties of the antennas are driven by intramolecular chromophoric stacking interactions that, upon forcing the a-helix to fold on itself, annul any effects deriving from the programming of the spatial arrangement of the receptor sides in the peptide backbone.
Controlling the Functional Properties of Oligothiophene Crystalline Nano/Microfibers via Tailoring of the Self-Assembling Molecular Precursors
Francesca Di Maria, Mattia Zangoli, Massimo Gazzano, Eduardo Fabiano, Denis Gentili, Alberto Zanelli, Andrea Fermi, Giacomo Bergamini, Davide Bonifazi, Andrea Perinot, Mario Caironi, Raffaello Mazzaro, Vittorio Morandi, Giuseppe Gigli, Andrea Liscio, Giovanna Barbarella
Journal
Adv. Funct. Mater.
Date
08/2018
Adv. Funct. Mater.
08/2018
Controlling the Functional Properties of Oligothiophene Crystalline Nano/Microfibers via Tailoring of the Self-Assembling Molecular Precursors
Francesca Di Maria, Mattia Zangoli, Massimo Gazzano, Eduardo Fabiano, Denis Gentili, Alberto Zanelli, Andrea Fermi, Giacomo Bergamini, Davide Bonifazi, Andrea Perinot, Mario Caironi, Raffaello Mazzaro, Vittorio Morandi, Giuseppe Gigli, Andrea Liscio, Giovanna Barbarella
Adv. Funct. Mater.
8 Aug 2018
Abstract
Oligothiophenes are -conjugated semiconducting and fluorescent molecules whose self-assembly properties are widely investigated for application in organic electronics, optoelectronics, biophotonics, and sensing. Here an approach to the preparation of crystalline oligothiophene nano/microfibers is reported based on the use of a sulfur overrich quaterthiophene building block, T4S4 , containing in its covalent network all the information needed to promote the directional, - stacking-driven, self-assembly of Y-T4S4-Y oligomers into fibers with hierarchical supramolecular arrangement from nano- to microscale. It is shown that when Y varies from unsubstituted thiophene to thiophene substituted with electron-withdrawing groups, a wide redistribution of the molecular electronic charge takes place without substantially affecting the aggregation modalities of the oligomer. In this way, a structurally comparable series of fibers is obtained having progressively varying optical properties, redox potentials, photoconductivity, and type of prevailing charge carriers (from p- to n-type). With the aid of density functional theory (DFT) calculations, combined with powder X-ray diffraction data, a model accounting for the growth of the fibers from molecular to nano- and microscale is proposed.
Oxygen-Doped Zig-Zag Molecular Ribbons
Andrey Berezin, Nicolas Biot, Tommaso Battisti, Davide Bonifazi
Journal
Angew. Chem.-Int. Edit.
Date
07/2018
Angew. Chem.-Int. Edit.
07/2018
Oxygen-Doped Zig-Zag Molecular Ribbons
Andrey Berezin, Nicolas Biot, Tommaso Battisti, Davide Bonifazi
Angew. Chem.-Int. Edit.
16 Jul 2018
Abstract
The synthesis of a zig-zag oxygen-doped molecular rhombic ribbon has been achieved. This includes oxidative C-C and C-O bond formations that allowed the stepwise elongation and planarization of an oxa-congener of 2,7-periacenoacene. X-ray diffraction analysis corroborated the flat structure and the zig-zag topology of the O-doped edges. Photophysical and electrochemical investigations showed that the extension of the peri-xanthenoxanthene (PXX) into the molecular ribbon induces a noticeable shrinking of the molecular band gap devised by a rising of the HOMO energy level, a desirable property for p-type organic semiconductors.
BN-Patterning of Metallic Substrates through Metal Coordination of Decoupled Borazines
Martin Schwarz, Manuela Garnica, Francesco Fasano, Nicola Demitri, Davide Bonifazi, Willi Auwaerter
Journal
Chem.-Eur. J.
Date
07/2018
Chem.-Eur. J.
07/2018
BN-Patterning of Metallic Substrates through Metal Coordination of Decoupled Borazines
Martin Schwarz, Manuela Garnica, Francesco Fasano, Nicola Demitri, Davide Bonifazi, Willi Auwaerter
Chem.-Eur. J.
5 Jul 2018
Abstract
We report on the synthesis of pyridine-terminated borazine derivatives, their molecular self-assembly as well as the electronic properties investigated on silver and copper surfaces by means of scanning tunneling microscopy and X-ray photoelectron spectroscopy. The introduction of pyridine functionalities allows us to achieve distinct supramolecular architectures with control of the interdigitation of the molecules by surface templating. On silver surfaces, the borazine derivatives arrange in a dense-packed hexagonal structure through van der Waals and H-bonding interactions, whereas on Cu(111), the molecules undergo metal coordination. The porosity and coordination symmetry of the reticulated structure depends on the stoichiometric ratio between copper adatoms and the borazine ligands, permitting an unusual three-fold coordinated Cu-pyridyl network. Finally, spectroscopy measurements indicate that the borazine core is electronically decoupled from the metallic substrate. We thus demonstrate that BNC-containing molecular units can be integrated into stable metal-coordination architectures on surfaces, opening pathways to patterned, BN-doped sheets with specific functionalities, for example, regarding the adsorption of polar guest gases.
Magnetic shepherding of nanocatalysts through hierarchically-assembled Fe-filled CNTs hybrids
Michele Melchionna, Alessandro Beltram, Antoine Stopin, Tiziano Montini, Rhys W. Lodge, Andrei N. Khlobystov, Davide Bonifazi, Maurizio Prato, Paolo Fornasiero
Journal
Appl. Catal. B-Environ.
Date
07/2018
Appl. Catal. B-Environ.
07/2018
Magnetic shepherding of nanocatalysts through hierarchically-assembled Fe-filled CNTs hybrids
Michele Melchionna, Alessandro Beltram, Antoine Stopin, Tiziano Montini, Rhys W. Lodge, Andrei N. Khlobystov, Davide Bonifazi, Maurizio Prato, Paolo Fornasiero
Appl. Catal. B-Environ.
5 Jul 2018
Abstract
Mechanically robust, chemically stable and electronically active carbon nanotubes (CNTs) are widely used as supports in catalysis. Synergistic effects between CNT and the active phase critically depend on the homogeneity of the carbon/inorganic interface, whose assembly is difficult to achieve without admixtures of free-standing inorganic matrix. Here we show that Fe-filled CNTs, employed as nanocatalyst supports, allow a facile preparation of highly pure and uniform CNT/nanocatalyst materials, by taking advantage of magnetic separation from poorly-defined components (e.g. aggregates of inorganic nanocatalysts). The higher homogeneity translates into higher catalytic activity in two industrially important processes: the photocatalytic hydrogen production and the water-gas shift reaction, WGSR (increase of similar to 48% activity for the former and up to similar to 45% for the latter as compared to catalysts isolated by standard filtration). In addition, the magnetic Fe core in the nanotubes enables effective separation and re-use of the nanocatalyst without loss of activity. This study demonstrates significant potential of magnetic CNTs as next generation of sustainable catalyst supports that can improve production of hydrogen and reduce the use of precious metals.
Kinked silicon nanowires-enabled interweaving electrode configuration for lithium-ion batteries
Georgiana Sandu, Michael Coulombier, Vishank Kumar, Hailu G. Kassa, Ionel Avram, Ran Ye, Antoine Stopin, Davide Bonifazi, Jean-Francois Gohy, Philippe Leclere, Xavier Gonze, Thomas Pardoen, Alexandru Vlad, Sorin Melinte
Journal
Sci Rep
Date
06/2018
Sci Rep
06/2018
Kinked silicon nanowires-enabled interweaving electrode configuration for lithium-ion batteries
Georgiana Sandu, Michael Coulombier, Vishank Kumar, Hailu G. Kassa, Ionel Avram, Ran Ye, Antoine Stopin, Davide Bonifazi, Jean-Francois Gohy, Philippe Leclere, Xavier Gonze, Thomas Pardoen, Alexandru Vlad, Sorin Melinte
Sci Rep
28 Jun 2018
Abstract
A tri-dimensional interweaving kinked silicon nanowires (k-SiNWs) assembly, with a Ni current collector co-integrated, is evaluated as electrode configuration for lithium ion batteries. The large-scale fabrication of k-SiNWs is based on a procedure for continuous metal assisted chemical etching of Si, supported by a chemical peeling step that enables the reuse of the Si substrate. The kinks are triggered by a simple, repetitive etch-quench sequence in a HF and H2O2-based etchant. We find that the interlocking frameworks of k-SiNWs and multi-walled carbon nanotubes exhibit beneficial mechanical properties with a foam-like behavior amplified by the kinks and a suitable porosity for a minimal electrode deformation upon Li insertion. In addition, ionic liquid electrolyte systems associated with the integrated Ni current collector repress the detrimental effects related to the Si-Li alloying reaction, enabling high cycling stability with 80% capacity retention (1695 mAh/g(Si)) after 100 cycles. Areal capacities of 2.42 mAh/cm(2) (1276 mAh/g(electrode)) can be achieved at the maximum evaluated thickness (corresponding to 1.3 mg(Si)/cm(2)). This work emphasizes the versatility of the metal assisted chemical etching for the synthesis of advanced Si nanostructures for high performance lithium ion battery electrodes.
Experimental Study of the Structural Effect on the Nanosecond Nonlinear Optical Response of O-Doped Polycyclic Aromatic Hydrocarbons
Ioannis Papadakis, Zoi Bouza, Aristeidis Stathis, Ioannis Orfanos, Stelios Couris, Tanja Miletic, Davide Bonifazi
Journal
J. Phys. Chem. A
Date
06/2018
J. Phys. Chem. A
06/2018
Experimental Study of the Structural Effect on the Nanosecond Nonlinear Optical Response of O-Doped Polycyclic Aromatic Hydrocarbons
Ioannis Papadakis, Zoi Bouza, Aristeidis Stathis, Ioannis Orfanos, Stelios Couris, Tanja Miletic, Davide Bonifazi
J. Phys. Chem. A
14 Jun 2018
Abstract
The nonlinear optical response of some O-doped polycyclic aromatic hydrocarbons (PAHs) is systematically investigated in the present work aiming to understand the influence of structural effects on their nonlinear optical response. In that view, the third-order nonlinear optical properties of these PAHs were measured under 4 ns visible (532 nm) and infrared (1064 nm) laser excitation. The O-doped PAHs were found to exhibit large saturable absorption and negative sign nonlinear refraction under visible excitation, increasing both with the addition of naphthalene units and with the number of O atoms. Their nonlinear optical response was found to be negligible under infrared excitation. Similar measurements performed on thin films of these PAHs have shown that they maintain their large nonlinear optical response even in the solid state, confirming their high potential for optoelectronic and photonic applications.
Programming Recognition Arrays through Double Chalcogen-Bonding Interactions
Nicolas Biot, Davide Bonifazi
Journal
Chem.-Eur. J.
Date
04/2018
Chem.-Eur. J.
04/2018
Programming Recognition Arrays through Double Chalcogen-Bonding Interactions
11 Apr 2018
Abstract
In this work, we have programmed and synthesized a recognition motif constructed around a chalcogenazolo-pyridine scaffold (CGP) that, through the formation of frontal double chalcogen-bonding interactions, associates into dimeric EX-type complexes. The reliability of the double chalcogen-bonding interaction has been shown at the solid-state by X-ray analysis, depicting the strongest recognition persistence for a Te-congener. The high recognition fidelity, chemical and thermal stability and easy derivatization at the 2-position makes CGP a convenient motif for constructing supramolecular architectures through programmed chalcogen-bonding interactions.
Customizing Photoredox Properties of PXX-based Dyes through Energy Level Rigid Shifts of Frontier Molecular Orbitals
Andrea Sciutto, Andrea Fermi, Andrea Folli, Tommaso Battisti, Joseph M. Beames, Damien M. Murphy, Davide Bonifazi
Journal
Chem.-Eur. J.
Date
03/2018
Chem.-Eur. J.
03/2018
Customizing Photoredox Properties of PXX-based Dyes through Energy Level Rigid Shifts of Frontier Molecular Orbitals
Andrea Sciutto, Andrea Fermi, Andrea Folli, Tommaso Battisti, Joseph M. Beames, Damien M. Murphy, Davide Bonifazi
Chem.-Eur. J.
20 Mar 2018
Abstract
Here we describe the synthesis of electron-rich PXX derivatives in which the energy levels of the excited states have been rigidly shifted through the insertion of imide groups. This has allowed the development of a new series of oxygen-doped photoredox-active chromophores with improved oxidizing and reducing properties. Capitalizing on the dehalogenation of organic halides as a model reaction, we could investigate the photooxidative and photoreductive potential of these molecules in model chemical transformations. Depending on the substrate, solvent and dye the reaction mechanism can follow different paths. This prompted us to consider the first chemoselective transformation protocol, in which two different C-Br bonds could be chemoselectively reacted through the sequential photoactivation of two different colorants.
Direction-dependent secondary bonds and their stepwise melting in a uracil-based molecular crystal studied by infrared spectroscopy and theoretical modeling
Zsolt Szekrenyes, Peter R. Nagy, Gyorgy Tarczay, Laura Maggini, Davide Bonifazi, Katalin Kamaras
Journal
Chem. Phys. Lett.
Date
01/2018
Chem. Phys. Lett.
01/2018
Direction-dependent secondary bonds and their stepwise melting in a uracil-based molecular crystal studied by infrared spectroscopy and theoretical modeling
Zsolt Szekrenyes, Peter R. Nagy, Gyorgy Tarczay, Laura Maggini, Davide Bonifazi, Katalin Kamaras
Chem. Phys. Lett.
1 Jan 2018
Abstract
Three types of supramolecular interactions are identified in the three crystallographic directions in crystals of 1,4-bis[(1-hexylurac-6-yl) ethynyl] benzene, a uracil-based molecule with a linear backbone. These three interactions, characterized by their strongest component, are: intermolecular double H-bonds along the molecular axis, London dispersion interaction of hexyl chains connecting these linear assemblies, and p-p stacking of the aromatic rings perpendicular to the molecular planes. On heating, two transitions happen, disordering of hexyl chains at 473 K, followed by H-bond melting at 534 K. The nature of the bonds and transitions was established by matrix-isolation and temperature-dependent infrared spectroscopy and supported by theoretical computations. (C) 2017 Elsevier B.V. All rights reserved.
2017
Title
Toward Fractioning of Isomers through Binding-Induced Acceleration of Azobenzene Switching
Rosaria Vulcano, Paolo Pengo, Simone Velari, Johan Wouters, Alessandro De Vita, Paolo Tecilla, Davide Bonifazi
DOI: 10.1021/jacs.7b09568
Journal
J. Am. Chem. Soc.
Date
12/2017
J. Am. Chem. Soc.
12/2017
Toward Fractioning of Isomers through Binding-Induced Acceleration of Azobenzene Switching
Rosaria Vulcano, Paolo Pengo, Simone Velari, Johan Wouters, Alessandro De Vita, Paolo Tecilla, Davide Bonifazi
DOI: 10.1021/jacs.7b09568
J. Am. Chem. Soc.
20 Dec 2017
Abstract
The E/Z isomerization process of a uracil-azobenzene derivative in which the nucleobase is conjugated to a phenyldiazene tail is studied in view of its ability to form triply H-bonded complexes with a suitably complementary 2,6-diacetyla-mino-4-pyridine ligand. UV-vis and H-1 NMR investigations of the photochemical and thermal isomerization kinetics show that the thermal Z -> E interconversion is 4-fold accelerated upon formation of the H-bonded complex. DFT calculations show that the formation of triple H-bonds triggers a significant elongation of the N = N double bond, caused by an increase of its pi(g)* antibonding character. This results in a reduction of the N = N torsional barrier and thus in accelerated thermal Z -> E isomerization. Combined with light-controlled E -> Z isomerization, this enables controllable fractional tuning of the two configurational isomers.
Stereospecific Winding of Polycyclic Aromatic Hydrocarbons into Trinacria Propellers
Dario Mosca, Antoine Stopin, Johan Wouters, Nicola Demitri, Davide Bonifazi
Journal
Chem.-Eur. J.
Date
11/2017
Chem.-Eur. J.
11/2017
Stereospecific Winding of Polycyclic Aromatic Hydrocarbons into Trinacria Propellers
Dario Mosca, Antoine Stopin, Johan Wouters, Nicola Demitri, Davide Bonifazi
Chem.-Eur. J.
2 Nov 2017
Abstract
The stereospecific trimerization of enantiomerically pure binaphthols with hexakis(bromomethyl)benzene gives access in one step to enantiomerically pure molecular propellers, in which three binaphthyl rings are held together with dioxecine rings. X-ray diffraction analysis revealed that three out the six naphthyl moieties are folded in a (EF)(3)-type arrangement held by three intramolecular C-H center dot center dot center dot pi interactions. This slips outward the three remaining naphthyl rings in a blade-like fashion, just like in three-folded propeller components. This peculiar conformation shows striking similarity to the mythological Sicilian symbol of Trinacria, from which the name trinacria propeller derives. The propeller conformation is also preserved in chlorinated solutions, as displayed by the presence of a peak at 4.7ppm typical of an aromatic proton resonance engaged in a C-H center dot center dot center dot pi interaction. The denaturation of the propeller-like conformation is obtained at high temperature, corresponding to activation energy for the ring inversion of ca. 18.2kcalmol(-1). Notably, halide-functionalized molecular propellers exposing I-atoms at the leading and trailing edges could be prepared stereo- and regiospecifically by choosing the relevant iodo-bearing BINOL derivative.
A Twisted Bay-Substituted Quaterrylene Phosphorescing in the NIR Spectral Region
Tanja Miletic, Andrea Fermi, Ioannis Papadakis, Ioannis Orfanos, Nikolaos Karampitsos, Aggelos Avramopoulos, Nicola Demitri, Federica De Leo, Simon J. A. Pope, Manthos G. Papadopoulos, Stelios Couris, Davide Bonifazi
Journal
Helv. Chim. Acta
Date
11/2017
Helv. Chim. Acta
11/2017
A Twisted Bay-Substituted Quaterrylene Phosphorescing in the NIR Spectral Region
Tanja Miletic, Andrea Fermi, Ioannis Papadakis, Ioannis Orfanos, Nikolaos Karampitsos, Aggelos Avramopoulos, Nicola Demitri, Federica De Leo, Simon J. A. Pope, Manthos G. Papadopoulos, Stelios Couris, Davide Bonifazi
Helv. Chim. Acta
1 Nov 2017
Abstract
The preparation of the first soluble quaterrylene derivative featuring peripheral tert-butyl substituents and sterically hindering, core-anchored triflate groups has been achieved. This involves a facile synthetic route based on an oxidative coupling of perylene precursors in the presence of H2O2 as oxidant. The steric hindrance between the TfO substituents at the central bay position of the quaterrylene board triggers a strong deformation of the central perylene planarity, which forces the quaterrylene platform to adopt a twisted geometry as shown by X-ray analysis. Exceptionally, photophysical investigations show that the core-twisted quaterrylene phosphoresces in the NIR spectral region at 1716 nm. Moreover, third-order nonlinear optical measurements on solutions and thin film containing the relevant molecule showed very large second hyperpolarizability values, as predicted by theoretical calculations at the CAM-B3LYP/6-31G** level of theory, making this material very appealing for photonic applications.
Renaissance of an Old Topic: From Borazines to BN-doped Nanographenes
Maria Mercedes Lorenzo-Garcia, Davide Bonifazi
Journal
Chimia
Date
09/2017
Chimia
09/2017
Renaissance of an Old Topic: From Borazines to BN-doped Nanographenes
27 Sep 2017
Abstract
Graphene is one of the leading materials in todays science, but the lack of a band gap limits its application to replace semiconductors in optoelectronic devices. To overcome this limitation, the replacement of C=C bonds by isostructural and isoelectronic bonds is emerging as an effective strategy to open a band gap in monoatomic graphene layers. First prepared by Stock and Pohland in 1926, borazine is the isoelectronic and isostructural inorganic analogue of benzene, where the C=C bonds are replaced by B N couples. The strong polarity of the BN bonds widens the molecular HOMO LUMO gap, imparting strong UV-emission/absorption and electrical insulating properties. These properties make borazine a valuable molecular scaffold to be inserted as doping units in graphitic-based carbon materials to tailor a relevant band gap. It is with this objective that we became interested in the development of new synthetic organic methodologies to gain access to functionalized borazine derivatives. In particular, we have described the synthesis of borazine derivatives that, featuring aryl substituents at the B-centers bearing ortho-functionalities, are exceptionally stable against hydrolysis. Building on these structural motifs, we prepared hybrid BN-doped polyphenylene nanostructures featuring controlled doping patterns, both as dosage and orientation. Finally, exploiting the Friedel-Craft electrophilic aromatic substitution, we could develop the first rational synthesis of the first soluble hexa-peri-hexabenzoborazinocoronene and measured its optoelectronic properties, showing a widening of its gap compared to its full-carbon congener.
Unfolding IGDQ Peptides for Engineering Motogenic Interfaces
Federica De Leo, Riccardo Marega, Valentina Corvaglia, Rodolfo Tondo, Matteo Lo Cicero, Simone Silvestrini, Davide Bonifazi
Journal
Langmuir
Date
08/2017
Langmuir
08/2017
Unfolding IGDQ Peptides for Engineering Motogenic Interfaces
Federica De Leo, Riccardo Marega, Valentina Corvaglia, Rodolfo Tondo, Matteo Lo Cicero, Simone Silvestrini, Davide Bonifazi
Langmuir
1 Aug 2017
Abstract
Extracellular matrix (ECM)-mimicking surfaces are pivotal tools in understanding adherent cell physiopathology. In this sense, we have recently reported on a discrete set of ECM-mimicking SAMs, among which only those exposing IGDQ peptide-alkanethiols sustain the adhesion of MDA-MB-231 cells by triggering FAK phosphorylation and peculiarly induce the migration of individual cancer cells on the subcentimeter scale. Starting from the experimentally observed relationship among the SAM composition, organization, and biological response, a systematic computational characterization aided in pinpointing the atomistic details through which specific composition and organization achieve the desired biological responsiveness. Specifically, the solvent, number and type of peptides, and presence or absence of surface fillers were accurately considered, creating representative model SAMs simulated by means of classical molecular dynamics (MD) with a view toward unravelling the experimental evidence, revealing how the conformational and structural features of these substrates dictate the specific motogenic responses. Through complementary experimental and computational investigations, it clearly emerges that there exists a distinct and precise mutual interaction among IGDQ-peptides, the surface fillers, and Au, which controls the structural properties of the ECM-mimicking SAMs and thus their motogenic potential.
Borazino-Doped Polyphenylenes
Davide Marinelli, Francesco Fasano, Btissam Najjari, Nicola Demitri, Davide Bonifazi
DOI: 10.1021/jacs.7b01477
Journal
J. Am. Chem. Soc.
Date
04/2017
J. Am. Chem. Soc.
04/2017
Borazino-Doped Polyphenylenes
Davide Marinelli, Francesco Fasano, Btissam Najjari, Nicola Demitri, Davide Bonifazi
DOI: 10.1021/jacs.7b01477
J. Am. Chem. Soc.
19 Apr 2017
Abstract
The divergent synthesis of two series of borazino-doped polyphenylenes, in which one or more aryl units are replaced by borazine rings, is reported for the first time, taking advantage of the decarbonylative [4 + 2] Diels-Alder cycloaddition reaction between ethynyl and tetraphenylcyclopentadienone derivatives. Because of the possibility of functionalizing the borazine core with different groups on the, aryl substituents at the N and B atoms of the borazino core, we have prepared borazino-doped polyphenylenes featuring different doping dosages and orientations. To achieve this, two molecular modules were prepared: a core and a branching unit. Depending on the chemical natures of the central aromatic module and the reactive group, each covalent combination of the modules yields one exclusive doping pattern. By means of this approach, three- and hexa-branched hybrid polyphenylenes featuring controlled orientations and dosages of the doping B3N3 rings have been prepared. Detailed photophysical investigations showed that as the doping dosage is increased, the strong luminescent signal is progressively reduced. This suggests that the presence of the B3N3 rings engages additional deactivation pathways, possibly involving excited states with an increasing charge-separated character that are restricted in the full-carbon analogues. Notably, a strong effect of the orientational doping on the fluorescence quantum yield was observed for those hybrid polyphenylene structures featuring low doping dosages. Finally, we showed that Cu-catalyzed 1,3-dipolar cycloaddition is also chemically compatible with the BN core, further endorsing the inorganic benzene as a versatile aromatic scaffold for engineering of molecular materials with tailored and exploitable optoelectronic properties.
Synthesis and Optoelectronic Properties of Hexa-peri-hexabenzoborazinocoronene
Jacopo Dosso, Jonathan Tasseroul, Francesco Fasano, Davide Marinelli, Nicolas Biot, Andrea Fermi, Davide Bonifazi
Journal
Angew. Chem.-Int. Edit.
Date
04/2017
Angew. Chem.-Int. Edit.
04/2017
Synthesis and Optoelectronic Properties of Hexa-peri-hexabenzoborazinocoronene
Jacopo Dosso, Jonathan Tasseroul, Francesco Fasano, Davide Marinelli, Nicolas Biot, Andrea Fermi, Davide Bonifazi
Angew. Chem.-Int. Edit.
10 Apr 2017
Abstract
The first rational synthesis of a BN-doped coronene derivative in which the central benzene ring has been replaced by a borazine core is described. This includes six C-C ring-closure steps that, through intramolecular Friedel-Crafts-type reactions, allow the stepwise planarization of the hexaarylborazine precursor. UV/Vis absorption, emission, and electrochemical investigations show that the introduction of the central BN core induces a dramatic widening of the HOMO-LUMO gap and an enhancement of the blue-shifted emissive properties with respect to its all-carbon congener.
Versatile Self-Adapting Boronic Acids for H-Bond Recognition: From Discrete to Polymeric Supramolecules
Irene Georgiou, Simon Kervyn, Alexandre Rossignon, Federica De Leo, Johan Wouters, Gilles Bruylants, Davide Bonifazi
DOI: 10.1021/jacs.6b11362
Journal
J. Am. Chem. Soc.
Date
02/2017
J. Am. Chem. Soc.
02/2017
Versatile Self-Adapting Boronic Acids for H-Bond Recognition: From Discrete to Polymeric Supramolecules
Irene Georgiou, Simon Kervyn, Alexandre Rossignon, Federica De Leo, Johan Wouters, Gilles Bruylants, Davide Bonifazi
DOI: 10.1021/jacs.6b11362
J. Am. Chem. Soc.
22 Feb 2017
Abstract
Because of the peculiar dynamic covalent reactivity of boronic acids to form tetraboronate derivatives, interest in using their aryl derivatives in materials science and supramolecular chemistry has risen. Nevertheless, their ability to form H-bonded complexes has been only marginally touched. Herein we report the first solution and solid-state binding studies of the first double-H-bonded DD-AA.-type complexes of a series of aromatic boronic acids that adopt a syn-syn conformation with suitable complementary H-bonding acceptor partners. The first determination of the association constant (K-a) of ortho-substituted boronic acids in solution showed that K-a for 1:1 association is in the range between 300 and 6900 M-1. Crystallization of dimeric 1:1 and trimeric 1:2 and 2:1 complexes enabled an in-depth examination of these complexes in the solid state, proving the selection of the B(OH)(2) syn syn conformer through a pair of frontal H-bonds with the relevant AA partner. Non-ortho-substituted boronic acids result in flat complexes. On the other hand, sterically demanding analogues bearing ortho substituents strive to retain their recognition properties by rotation of the ArB(OH)(2) moiety, forming T-shaped complexes. Solid-state studies of a diboronic acid and a tetraazanaphthacene provided for the first time the formation of a supramolecular H-bonded polymeric ribbon. On the basis of the conformational dynamicity of the B(OH)(2) functional group, it is expected that these findings will also open new possibilities in metal-free catalysis or organic crystal engineering, where double H -bonding donor boronic acids could act as suitable organocatalysts or templates for the development of functional materials with tailored organizational properties.
Tailoring Colors by O Annulation of Polycyclic Aromatic Hydrocarbons
Tanja Miletic, Andrea Fermi, Ioannis Orfanos, Aggelos Avramopoulos, Federica De Leo, Nicola Demitri, Giacomo Bergamini, Paola Ceroni, Manthos G. Papadopoulos, Stelios Couris, Davide Bonifazi
Journal
Chem.-Eur. J.
Date
02/2017
Chem.-Eur. J.
02/2017
Tailoring Colors by O Annulation of Polycyclic Aromatic Hydrocarbons
Tanja Miletic, Andrea Fermi, Ioannis Orfanos, Aggelos Avramopoulos, Federica De Leo, Nicola Demitri, Giacomo Bergamini, Paola Ceroni, Manthos G. Papadopoulos, Stelios Couris, Davide Bonifazi
Chem.-Eur. J.
1 Feb 2017
Abstract
The synthesis of O-doped polyaromatic hydro-carbons in which two polycyclic aromatic hydrocarbon sub units are bridged through one or two O atoms has been achieved. This includes high-yield ring-closure key steps that, depending on the reaction conditions, result in the formation of furanyl or pyranopyranyl linkages through intramolecular C-O bond formation. Comprehensive photophysical measurements in solution showed that these compounds have exceptionally high emission yields and tunable absorption properties throughout the UV/Vis spectral region. Electrochemical investigations showed that in all cases O annulation increases the electron-donor capabilities by raising the HOMO energy level, whereas the LUMO energy level is less affected. Moreover, third-order nonlinear optical (NLO) measurements on solutions or thin films containing the dyes showed very good values of the second hyperpolarizability. Importantly, poly(methyl methacrylate) films containing the pyranopyranyl derivatives exhibited weak linear absorption and NLO absorption compared to the nonlinearity and NLO refraction, respectively, and thus revealed them to be exceptional organic materials for photonic devices.
2016
Title
LET-dependent radiosensitization effects of gold nanoparticles for proton irradiation
Sha Li, Sebastien Penninckx, Linda Karmani, Anne-Catherine Heuskin, Kassandra Watillon, Riccardo Marega, Valentina Corvaglia, Geraldine Genard, Bernard Gallez, Olivier Feron, Philippe Martinive, Davide Bonifazi, Carine Michiels, Stephane Lucas
Journal
Nanotechnology
Date
11/2016
Nanotechnology
11/2016
LET-dependent radiosensitization effects of gold nanoparticles for proton irradiation
Sha Li, Sebastien Penninckx, Linda Karmani, Anne-Catherine Heuskin, Kassandra Watillon, Riccardo Marega, Valentina Corvaglia, Geraldine Genard, Bernard Gallez, Olivier Feron, Philippe Martinive, Davide Bonifazi, Carine Michiels, Stephane Lucas
Nanotechnology
15 Nov 2016
Abstract
The development of new modalities and protocols is of major interest to improve the outcome of cancer treatment. Given the appealing physical properties of protons and the emerging evidence of biological relevance of the use of gold nanoparticles (GNPs), the radiosensitization effects of GNPs (5 or 10nm) have been investigated in vitro in combination with a proton beam of different linear energy transfer (LET). After the incubation with GNPs for 24 h, nanoparticles were observed in the cytoplasm of A431 cells exposed to 10 nm GNPs, and in the cytoplasm as well as the nucleus of cells exposed to 5 nm GNPs. Cell uptake of 0.05 mg ml(-1) of GNPs led to 0.78 pg Au/cell and 0.30 pg Au/cell after 24 h incubation for 10 and 5 nm GNPs respectively. A marked radiosensitization effect of GNPs was observed with 25 keV mu m(-1) protons, but not with 10 keV mu m(-1) protons. This effect was more pronounced for 10 nm GNPs than for 5 nm GNPs. By using a radical scavenger, a major role of reactive oxygen species in the amplification of the death of irradiated cell was identified. All together, these results open up novel perspectives for using high-Z metallic NPs in protontherapy.
Fast Targeting and Cancer Cell Uptake of Luminescent Antibody-Nanozeolite Bioconjugates
Riccardo Marega, Eko Adi Prasetyanto, Carine Michiels, Luisa De Cola, Davide Bonifazi
Journal
Small
Date
10/2016
Small
10/2016
Fast Targeting and Cancer Cell Uptake of Luminescent Antibody-Nanozeolite Bioconjugates
Riccardo Marega, Eko Adi Prasetyanto, Carine Michiels, Luisa De Cola, Davide Bonifazi
Small
19 Oct 2016
Abstract
Understanding the targeted cellular uptake of nanomaterials is an essential step to engineer and program functional and effective biomedical devices. In this respect, the targeting and ultrafast uptake of zeolite nanocrystals functionalized with Cetuximab antibodies (Ctxb) by cells overexpressing the epidermal growth factor receptor are described here. Biochemical assays show that the cellular uptake of the bioconjugate in the targeted cancer cells already begins 15 min after incubation, at a rate around tenfold faster than that observed in the negative control cells. These findings further show the role of Ctxb exposed at the surfaces of the zeolite nanocrystals in mediating the targeted and rapid cellular uptake. By using temperature and pharmacological inhibitors as modulators of the internalization pathways, the results univocally suggest a dissipative uptake mechanism of these nanomaterials, which seems to occur using different internalization pathways, according to the targeting properties of these nanocrystals. Owing to the ultrafast uptake process, harmless for the cell viability, these results further pave the way for the design of novel theranostic tools based on nanozeolites.
Covalently Functionalized SWCNTs as Tailored p-Type Dopants for Perovskite Solar Cells
Tanja Miletic, Eleonora Pavoni, Vanira Trifiletti, Aurora Rizzo, Andrea Listorti, Silvia Colella, Nicola Armaroli, Davide Bonifazi
Journal
ACS Appl. Mater. Interfaces
Date
10/2016
ACS Appl. Mater. Interfaces
10/2016
Covalently Functionalized SWCNTs as Tailored p-Type Dopants for Perovskite Solar Cells
Tanja Miletic, Eleonora Pavoni, Vanira Trifiletti, Aurora Rizzo, Andrea Listorti, Silvia Colella, Nicola Armaroli, Davide Bonifazi
ACS Appl. Mater. Interfaces
19 Oct 2016
Abstract
The covalent functionalization of (7,6)-enriched single walled carbon nanotubes (SWCNTs) with oligophenylenevinylene (OPV) moieties terminating with a dimethylamino group is proposed as an efficient way to enhance the affinity of CNTs with spiro-MeOTAD in perovskite-based solar cells. The evidence of SWCNTs functionalization and the degree of OPV substitution on SWCNTs are established from TGA, XPS, TEM, and Raman techniques. Our tailored doping materials afford photovoltaic performances in line with conventional Li doped spiro-MeOTAD, showing at the same time a significantly improved chemical stability of the perovskite component over time. Furthermore, the comparison of the photovoltaic performances with those obtained with nonfunctionalized SWCNTs suggest that the presence of the organic appends ensures highly reproducible PV performances. These results demonstrate the suitability of this functionalized SWCNT material as a valid doping agent for spiro-MeOTAD, representing a viable alternative to the conventional Li salt.
Polarization of Soft Materials through Magnetic Alignment of Polymeric Organogels under Low-Field Conditions
Antoine Stopin, Alexandre Rossignon, Masoumeh Keshavarz, Yasuhiro Ishida, Peter C. M. Christianen, Davide Bonifazi
Journal
Chem. Mat.
Date
10/2016
Chem. Mat.
10/2016
Polarization of Soft Materials through Magnetic Alignment of Polymeric Organogels under Low-Field Conditions
Antoine Stopin, Alexandre Rossignon, Masoumeh Keshavarz, Yasuhiro Ishida, Peter C. M. Christianen, Davide Bonifazi
Chem. Mat.
11 Oct 2016
Abstract
Through application of an external magnetic field upon jellification of poly(3-hydroxybutyric acid-co-3-hydroxyvaleric) acid (PHBV) polymer in different solvents, an anisotropic organogel is obtained. This material presents two alignment steps in an external magnetic field, in the liquid phase and during the jellification, both phenomena measured by magnetic field induced linear birefringence. Remarkably, the organogel developed in this study presents a strong level of birefringence, 80% of its maximum, in an external magnetic field as low as 2 T resulting from the magnetic alignment of the fibers of the material. This anisotropic material shows changes of absorbance upon rotation of a polarizer switching from transparent to opaque. In addition, its suprastructure does not influence the luminescent properties of encapsulated chromophores, allowing the formation of colored anisotropic materials.
Synthesis of Tertiary Enamides by Ag2CO3-Promoted Pd-Catalyzed Alkenylation of Acyclic Secondary Amides
Arnaud Delforge, Irene Georgiou, Adrian Kremer, Johan Wouters, Davide Bonifazi
Journal
Org. Lett.
Date
10/2016
Org. Lett.
10/2016
Synthesis of Tertiary Enamides by Ag2CO3-Promoted Pd-Catalyzed Alkenylation of Acyclic Secondary Amides
Arnaud Delforge, Irene Georgiou, Adrian Kremer, Johan Wouters, Davide Bonifazi
Org. Lett.
7 Oct 2016
Abstract
A Pd-catalyzed methodology for the preparation of tertiary enamides from acyclic secondary amides and bromo acrylates under mild reaction conditions has been developed using [Pd-2(dba)(3)], XantPhos, and Ag2CO3 as a base. The reaction occurs through a stereospecific metal-mediated oxidative-insertion mechanism.
Tailoring Large Pores of Porphyrin Networks on Ag(111) by MetalOrganic Coordination
Felix Bischoff, Yuanqin He, Knud Seufert, Daphne Stassen, Davide Bonifazi, Johannes V. Barth, Willi Auwaerter
Journal
Chem.-Eur. J.
Date
10/2016
Chem.-Eur. J.
10/2016
Tailoring Large Pores of Porphyrin Networks on Ag(111) by MetalOrganic Coordination
Felix Bischoff, Yuanqin He, Knud Seufert, Daphne Stassen, Davide Bonifazi, Johannes V. Barth, Willi Auwaerter
Chem.-Eur. J.
1 Oct 2016
Abstract
The engineering of nanoarchitectures to achieve tailored properties relevant for macroscopic devices is a key motivation of organometallic surface science. To this end, understanding the role of molecular functionalities in structure formation and adatom coordination is of great importance. In this study, the differences in formation of Cu-mediated metal-organic coordination networks based on two pyridyl- and cyano-bearing free-base porphyrins on Ag(111) are elucidated by use of low-temperature scanning tunneling microscopy (STM). Distinct coordination networks evolve via different pathways upon codeposition of Cu adatoms. The cyano-terminated module directly forms 2D porous networks featuring fourfold-coordinated Cu nodes. By contrast, the pyridyl species engage in twofold coordination with Cu and a fully reticulated 2D network featuring a pore size exceeding 3 nm(2) only evolves via an intermediate structure based on 1D coordination chains. The STM data and complementary Monte Carlo simulations reveal that these distinct network architectures originate from spatial constraints at the coordination centers. Cu adatoms are also shown to form two- and fourfold monoatomic coordination nodes with monotopic nitrogen-terminated linkers on the very same metal substrate-a versatility that is not achieved by other 3d transition metal centers but consistent with 3D coordination chemistry. This study discloses how specific molecular functionalities can be applied to tailor coordination architectures and highlights the potential of Cu as coordination center in such low-dimensional structures on surfaces.
Supramolecular Spangling, Crocheting, and Knitting of Functionalized Pyrene Molecules on a Silver Surface
Tobias Kaposi, Sushobhan Joshi, Tobias Hoh, Alissa Wiengarten, Knud Seufert, Matheusz Paszkiewicz, Florian Klappenberger, David Ecija, Luka Dordevic, Tomas Marangoni, Johannes V. Barth, Willi Auwaerter
Journal
ACS Nano
Date
08/2016
ACS Nano
08/2016
Supramolecular Spangling, Crocheting, and Knitting of Functionalized Pyrene Molecules on a Silver Surface
Tobias Kaposi, Sushobhan Joshi, Tobias Hoh, Alissa Wiengarten, Knud Seufert, Matheusz Paszkiewicz, Florian Klappenberger, David Ecija, Luka Dordevic, Tomas Marangoni, Johannes V. Barth, Willi Auwaerter
ACS Nano
1 Aug 2016
Abstract
Pyrenes, as photoactive polycyclic aromatic hydrocarbons (PAHs), represent promising modules for the bottom-up assembly of functional nanostructures. Here, we introduce the synthesis of a family of pyrene derivatives peripherally functionalized with pyridin-4-ylethynyl termini and comprehensively characterize their self-assembly abilities on a smooth Ag(111) support by scanning tunneling microscopy. By deliberate selection of number and geometric positioning of the pyridyl-terminated substituents, two-dimensional arrays, one-dimensional coordination chains, and chiral, porous kagome-type networks can be tailored. A comparison to phenyl-functionalized reference pyrenes, not supporting the self-assembly of ordered structures at low coverage, highlights the role of the pyridyl moieties for supramolecular crocheting and knitting. Furthermore, we demonstrate the selective spangling of pores in the two-dimensional pyrene assemblies by a distinct number of iodine atoms as guests by atomically resolved imaging and complementary X-ray photoelectron spectroscopy.
Biodistribution of 125I-labeled anti-endoglin antibody using SPELT/CT imaging: Impact of in vivo deiodination on tumor accumulation in mice
Linda Karmani, Philippe Leveque, Caroline Bouzin, Anne Bol, Marc Dieu, Stephan Walrand, Thierry Vander Borght, Olivier Feron, Vincent Gregoire, Davide Bonifazi, Carine Michiels, Stephane Lucas, Bernard Gallez
Journal
Nucl. Med. Biol.
Date
07/2016
Nucl. Med. Biol.
07/2016
Biodistribution of 125I-labeled anti-endoglin antibody using SPELT/CT imaging: Impact of in vivo deiodination on tumor accumulation in mice
Linda Karmani, Philippe Leveque, Caroline Bouzin, Anne Bol, Marc Dieu, Stephan Walrand, Thierry Vander Borght, Olivier Feron, Vincent Gregoire, Davide Bonifazi, Carine Michiels, Stephane Lucas, Bernard Gallez
Nucl. Med. Biol.
1 Jul 2016
Abstract
Introduction: Radiolabeled antibodies directed against endoglin (CD105) are promising tools for imaging and antiangiogenic cancer therapy. To validate iodinated antibodies as reliable tracers, we investigated the influence of the radiolabeling method (direct or indirect) on their in vivo stability. Methods: Anti-CD105 mAbs were radioiodinated directly using chloramine-T (I-125-anti-CD105-mAbs) or indirectly using D-KRYRR peptide as a linker (I-125-KRYRR-anti-CD105-mAbs). The biodistribution was studied in B16 tumor-bearing mice via SPECT/CT imaging. Results: Radioiodinated mAbs were stable in vitro. In vivo, thyroid showed the most important increase of uptake after 24 h for I-125-anti-CD105-mAbs (91.9 +/- 4.0%ID/ml) versus I-125-KRYRR-anti-CD105-mAbs (4.4 +/- 0.6%ID/ml). Tumor uptake of I-125-anti-CD105-mAbs (0.9 +/- 0.3%ID/ml) was significantly lower than that of (IKRYRR)-I-125-anti-CD105-mAbs (4.7 +/- 02%ID/ml). Conclusions: An accurate characterization of the in vivo stability of radioiodinated mAbs and the choice of an appropriate method for the radioiodination are required, especially for novel targets. The indirect radioiodination of internalizing anti-CD105 mAbs leads to more stable tracer by decreasing in vivo deiodination and improves the tumor retention of radioiodinated mAbs. Advances in knowledge and implications for patient care: To date, the only antiangiogenic antibody approved for clinical indications is bevacizumab. There is a need to develop more antibodies that have targets highly expressed on tumor endothelium. CD105 represents a promising marker of angiogenesis, but its therapeutic relevance in cancer needs to be further investigated. In this context, this study suggests the potential use of indirectly iodinated anti-CD105 mAbs for tumor imaging and for therapeutic purposes. (C) 2016 Elsevier Inc. All rights reserved.
Two-Dimensional Ketone-Driven Metal-Organic Coordination on Cu(111)
Ada Della Pia, Massimo Riello, James Lawrence, Daphne Stassen, Tim S. Jones, Davide Bonifazi, Alessandro De Vita, Giovanni Costantini
Journal
Chem.-Eur. J.
Date
06/2016
Chem.-Eur. J.
06/2016
Two-Dimensional Ketone-Driven Metal-Organic Coordination on Cu(111)
Ada Della Pia, Massimo Riello, James Lawrence, Daphne Stassen, Tim S. Jones, Davide Bonifazi, Alessandro De Vita, Giovanni Costantini
Chem.-Eur. J.
6 Jun 2016
Abstract
Two-dimensional metal-organic nanostructures based on the binding of ketone groups and metal atoms were fabricated by depositing pyrene-4,5,9,10-tetraone (PTO) molecules on a Cu(111) surface. The strongly electronegative ketone moieties bind to either copper adatoms from the substrate or codeposited iron atoms. In the former case, scanning tunnelling microscopy images reveal the development of an extended metal-organic supramolecular structure. Each copper adatom coordinates to two ketone ligands of two neighbouring PTO molecules, forming chains that are linked together into large islands through secondary van der Waals interactions. Deposition of iron atoms leads to a transformation of this assembly resulting from the substitution of the metal centres. Density functional theory calculations reveal that the driving force for the metal substitution is primarily determined by the strength of the ketone-metal bond, which is higher for Fe than for Cu. This second class of nanostructures displays a structural dependence on the rate of iron deposition.
Extended O-Doped Polycyclic Aromatic Hydrocarbons
Daphne Stassen, Nicola Demitri, Davide Bonifazi
Journal
Angew. Chem.-Int. Edit.
Date
05/2016
Angew. Chem.-Int. Edit.
05/2016
Extended O-Doped Polycyclic Aromatic Hydrocarbons
10 May 2016
Abstract
The synthesis of O-doped benzorylenes, in which peripheral carbon atoms have been replaced by oxygen atoms, has been achieved for the first time. This includes key high-yielding ring-closure steps which, through intramolecular C-O bond formation, allow stepwise planarization of oligonaphthalenes. Single-crystal X-ray diffraction showed that the tetraoxa derivative forms remarkable face-to-face pi-pi stacks in the solid state, a favorable solid-state arrangement for organic electronics.
Supramolecular Wiring of Benzo-1,3-chalcogenazoles through Programmed Chalcogen Bonding Interactions
Adrian Kremer, Andrea Fermi, Nicolas Biot, Johan Wouters, Davide Bonifazi
Journal
Chem.-Eur. J.
Date
04/2016
Chem.-Eur. J.
04/2016
Supramolecular Wiring of Benzo-1,3-chalcogenazoles through Programmed Chalcogen Bonding Interactions
Adrian Kremer, Andrea Fermi, Nicolas Biot, Johan Wouters, Davide Bonifazi
Chem.-Eur. J.
11 Apr 2016
Abstract
The high-yielding synthesis of 2-substituted benzo-1,3-tellurazoles and benzo-1,3-selenazoles through a dehydrative cyclization reaction has been reported, giving access to a large variety of benzo-1,3-chalcogenazoles. Exceptionally, these aromatic heterocycles proved to be very stable and thus very handy to form controlled solid-state organizations in which wire-like polymeric structures are formed through secondary NY bonding interactions (SBIs) engaging the chalcogen (Y=Se or Te) and nitrogen atoms. In particular, it has been shown that the recognition properties of the chalcogen centre at the solid state could be programmed by selectively barring one of its sigma-holes through a combination of electronic and steric effects exerted by the substituent at the 2-position. As predicted by the electrostatic potential surfaces calculated by quantum chemical modelling, the pyridyl groups revealed to be the stronger chalcogen bonding acceptors, and thus the best ligand candidate for programming the molecular organization at the solid state. In contrast, the thiophenyl group is an unsuitable substituent for establishing SBIs in this molecular system as it gives rise to chalcogen-chalcogen repulsion. The weaker chalcogen donor properties of the Se analogues trigger the formation of feeble NSe contacts, which are manifested in similar solid-state polymers featuring longer nitrogen-chalcogen distances.
Solvent-dependent moulding of porphyrin-based nanostructures: solid state, solution and on surface self-assembly
Luka Dordevic, Nicola Demitri, Davide Bonifazi
Journal
Supramol. Chem.
Date
03/2016
Supramol. Chem.
03/2016
Solvent-dependent moulding of porphyrin-based nanostructures: solid state, solution and on surface self-assembly
18 Mar 2016
Abstract
A novel porphyrin derivative 1.Zn was synthesised in order to mimic the self-assembly properties of natural light-harvesting antennas and its self-assembly behaviour in solution and in solid state were studied by NMR and X-Ray spectroscopies. The self-assembly of this molecule was triggered in apolar solvents and studied in solution by UV-Vis spectroscopy, suggesting it is able to form slipped face-to-face aggregates, or J-aggregates. The nanoscopic and microscopic morphology of the aggregates was elucidated by atomic force microscopy, revealing the formation of extended two-dimensional structures. [GRAPHICS] .
Unleashing Cancer Cells on Surfaces Exposing Motogenic IGDQ Peptides
Valentina Corvaglia, Riccardo Marega, Federica De Leo, Carine Michiels, Davide Bonifazi
Journal
Small
Date
01/2016
Small
01/2016
Unleashing Cancer Cells on Surfaces Exposing Motogenic IGDQ Peptides
Valentina Corvaglia, Riccardo Marega, Federica De Leo, Carine Michiels, Davide Bonifazi
Small
20 Jan 2016
Abstract
Thiolated peptides bearing the Ile-Gly-Asp (IGD) motif, a highly conserved sequence of fibronectin, are used for the preparation of anisotropic self-assembled monolayers (SAM gradients) to study the whole-population migratory behavior of metastatic breast cancer cells (MDA-MB-231 cells). Ile-Gly-Asp-Gln-(IGDQ)-exposing SAMs sustain the adhesion of MDA-MB-231 cells by triggering focal adhesion kinase phosphorylation, similarly to the analogous Gly-Arg-Gly-Asp-(GRGD)-terminating surfaces. However, the biological responses of different cell lines interfaced with the SAM gradients show that only those exposing the IGDQ sequence induce significant migration of MDA-MB-231 cells. In particular, the observed migratory behavior suggests the presence of cell subpopulations associated with a stationary or a migratory phenotype, the latter determining a considerable cell migration at the sub-cm length scale. These findings are of great importance as they suggest for the first time an active role of biological surfaces exposing the IGD motif in the multicomponent orchestration of cellular signaling involved in the metastatic progression.
2015
Title
Templated Chromophore Assembly by Dynamic Covalent Bonds
Lou Rocard, Andrey Berezin, Federica De Leo, Davide Bonifazi
Journal
Angew. Chem.-Int. Edit.
Date
12/2015
Angew. Chem.-Int. Edit.
12/2015
Templated Chromophore Assembly by Dynamic Covalent Bonds
Lou Rocard, Andrey Berezin, Federica De Leo, Davide Bonifazi
Angew. Chem.-Int. Edit.
21 Dec 2015
Abstract
Through the simultaneous use of three orthogonal dynamic covalent reactions, namely disulfide, boronate, and acyl hydrazone formation, we conceived a facile and versatile protocol to spatially organize tailored chromophores, which absorb in the blue, red, and yellow regions, on a preprogrammed a-helix peptide. This approach allowed the assembly of the dyes in the desired ratio and spacing, as dictated by both the relative positioning and distribution of the recognition units on the peptide scaffold. Steady-state UV/Vis absorption and emission studies suggest an energy transfer from the yellow and red donors to the blue acceptor. A molecular dynamics simulation supports the experimental findings that the helical structure is maintained after the assembly and the three dyes are confined in defined conformational spaces.
Walking Down the Chalcogenic Group of the Periodic Table: From Singlet to Triplet Organic Emitters
Adrian Kremer, Claudia Aurisicchio, Federica De Leo, Barbara Ventura, Johan Wouters, Nicola Armaroli, Andrea Barbieri, Davide Bonifazi
Journal
Chem.-Eur. J.
Date
10/2015
Chem.-Eur. J.
10/2015
Walking Down the Chalcogenic Group of the Periodic Table: From Singlet to Triplet Organic Emitters
Adrian Kremer, Claudia Aurisicchio, Federica De Leo, Barbara Ventura, Johan Wouters, Nicola Armaroli, Andrea Barbieri, Davide Bonifazi
Chem.-Eur. J.
19 Oct 2015
Abstract
The synthesis, X-ray crystal structures, ground- and excited-state UV/Vis absorption spectra, and luminescence properties of chalcogen-doped organic emitters equipped on both extremities with benzoxa-, benzothia-, benzoselena- and benzotellurazole (1(X) and 2(X)) moieties have been reported for the first time. The insertion of the four different chalcogen atoms within the same molecular skeleton enables the investigation of only the chalcogenic effect on the organisation and photophysical properties of the material. Detailed crystal-structure analyses provide evidence of similar packing for 2(O)-2(Se), in which the benzoazoles are engaged in - stacking and, for the heavier atoms, in secondary XX and XN bonding interactions. Detailed computational analysis shows that the arrangement is essentially governed by the interplay of van der Waals and secondary bonding interactions. Progressive quenching of the fluorescence and concomitant onset of phosphorescence features with gradually shorter lifetimes are detected as the atomic weight of the chalcogen heteroatom increases, with the tellurium-doped derivatives exhibiting only emission from the lowest triplet excited state. Notably, the phosphorescence spectra of the selenium and tellurium derivatives can be recorded even at room temperature; this is a very rare finding for fully organic emitters.
Biotechnological promises of Fe-filled CNTs for cell shepherding and magnetic fluid hyperthermia applications
Florent Pineux, Riccardo Marega, Antoine Stopin, Alessandro La Torre, Yann Garcia, Eamonn Devlin, Carine Michiels, Andrei N. Khlobystov, Davide Bonifazi
DOI: 10.1039/c5nr04824a
Journal
Nanoscale
Date
10/2015
Nanoscale
10/2015
Biotechnological promises of Fe-filled CNTs for cell shepherding and magnetic fluid hyperthermia applications
Florent Pineux, Riccardo Marega, Antoine Stopin, Alessandro La Torre, Yann Garcia, Eamonn Devlin, Carine Michiels, Andrei N. Khlobystov, Davide Bonifazi
DOI: 10.1039/c5nr04824a
Nanoscale
2 Oct 2015
Abstract
Fe-filled carbon nanotubes (Fe@CNTs) recently emerged as an effective class of hybrid nanoparticles for biotechnological applications, such as magnetic cell sorting and magnetic fluid hyperthermia. Aiming at studying the effects of both the Fe loading and the magnetocrystalline characteristics in these applications, we describe herein the preparation of Fe@CNTs containing different Fe phases that, upon functionalization with the antibody Cetuximab (Ctxb), allow the targeting of cancer cells. Our experimental findings reveal that an optimal Ctxb/Fe weight ratio of 1.2 is needed for efficient magnetic cell shepherding, whereas enhanced MFH-induced mortality (70 vs. 15%) can be reached with hybrids enriched in the coercive Fe3C phase. These results suggest that a synergistic effect between the Ab loading and the Fe distribution in each nanotube exists, for which the maximum shepherding and hyperthermia effects are observed when higher densities of Fe@CNTs featuring the more coercive phase are interfaced with the cells.
Boron-nitrogen doped carbon scaffolding: organic chemistry, self-assembly and materials applications of borazine and its derivatives
Davide Bonifazi, Francesco Fasano, M. Mercedes Lorenzo-Garcia, Davide Marinelli, Hamid Oubaha, Jonathan Tasseroul
DOI: 10.1039/c5cc06611e
Journal
Chem. Commun.
Date
09/2015
Chem. Commun.
09/2015
Boron-nitrogen doped carbon scaffolding: organic chemistry, self-assembly and materials applications of borazine and its derivatives
Davide Bonifazi, Francesco Fasano, M. Mercedes Lorenzo-Garcia, Davide Marinelli, Hamid Oubaha, Jonathan Tasseroul
DOI: 10.1039/c5cc06611e
Chem. Commun.
18 Sep 2015
Abstract
Discovered by Stock and Pohland in 1926, borazine is the isoelectronic and isostructural inorganic analogue of benzene, where the CQC bonds are substituted by B-N bonds. The strong polarity of such heteroatomic bonds widens the HOMO-LUMO gap of the molecule, imparting strong UV-emitting/absorption and electrical insulating properties. These properties make borazine and its derivatives valuable molecular scaffolds to be inserted as doping units in graphitic-based carbon materials to tailor their optoelectronic characteristics, and specifically their semiconducting properties. By guiding the reader through the most significant examples in the field, in this feature paper we describe the past and recent developments in the organic synthesis and functionalisation of borazine and its derivatives. These boosted the production of a large variety of tailored derivatives, broadening their use in optoelectronics, H-2 storage and supramolecular functional architectures, to name a few.
Carboxylated, Fe-Filled Multiwalled Carbon Nanotubes as Versatile Catalysts for O2 Reduction and H2 Evolution Reactions at Physiological pH
M. Victoria Bracamonte, Michele Melchionna, Antoine Stopin, Angela Giulani, Claudio Tavagnacco, Yann Garcia, Paolo Fornasiero, Davide Bonifazi, Maurizio Prato
Journal
Chem.-Eur. J.
Date
09/2015
Chem.-Eur. J.
09/2015
Carboxylated, Fe-Filled Multiwalled Carbon Nanotubes as Versatile Catalysts for O2 Reduction and H2 Evolution Reactions at Physiological pH
M. Victoria Bracamonte, Michele Melchionna, Antoine Stopin, Angela Giulani, Claudio Tavagnacco, Yann Garcia, Paolo Fornasiero, Davide Bonifazi, Maurizio Prato
Chem.-Eur. J.
1 Sep 2015
Abstract
The development of new electrocatalysts for the oxygen reduction reaction (ORR) and hydrogen evolution reaction (HER) at physiological pH is critical for several fields, including fuel cells and biological applications. Herein, the assembly of an electrode based on carboxyl-functionalised hydrophilic multiwalled carbon nanotubes (MWCNTs) filled with Fe phases and their excellent performance as electrocatalysts for ORR and HER at physiological pH are reported. The encapsulated Fe dramatically enhances the catalytic activity, and the graphitic shells play a double role of efficiently mediating the electron transfer to O-2 and H2O reactants and providing a cocoon that prevents uncontrolled Fe oxidation or leaching.
Interfacing proteins with graphitic nanomaterials: from spontaneous attraction to tailored assemblies
Federica De Leo, Alessandra Magistrato, Davide Bonifazi
DOI: 10.1039/c5cs00190k
Journal
Chem. Soc. Rev.
Date
07/2015
Chem. Soc. Rev.
07/2015
Interfacing proteins with graphitic nanomaterials: from spontaneous attraction to tailored assemblies
3 Jul 2015
Abstract
This critical review aims at giving insights on the spontaneous tendency of proteins and their constitutive parts to adsorb on graphitic nanomaterials (GNMs) through non-covalent interactions occurring at their interfaces. Specifically, it focuses on the theoretical and experimental studies carried out to comprehend in depth the forces ruling the adsorption processes of proteins on fullerene, carbon nanotubes and graphene. In a systematic way the reader is guided through the most recent and representative examples describing at the atomistic level of detail the structural modalities and the chemico-physical principles through which amino acids, polypeptides and folded proteins interact with GNMs surface, thereby taking into consideration the mutual effects of both protein structural complexity and nanomaterial topology. Based on their chemical and structural features, the study and understanding of the protein-nanomaterial interfaces can be exploited in the view of design and control the spontaneous formation of biologically-active hybrid materials for the development of new tailored applications in the field of sensing, nanomedicine and biochemistry.
Solvent Molding of Organic Morphologies Made of Supramolecular Chiral Polymers
Luka Dordevic, Tomas Marangoni, Tanja Miletic, Jenifer Rubio-Magnieto, John Mohanraj, Heinz Amenitsch, Dario Pasini, Nikos Liaros, Stelios Couris, Nicola Armaroli, Mathieu Surin, Davide Bonifazi
DOI: 10.1021/jacs.5b02448
Journal
J. Am. Chem. Soc.
Date
07/2015
J. Am. Chem. Soc.
07/2015
Solvent Molding of Organic Morphologies Made of Supramolecular Chiral Polymers
Luka Dordevic, Tomas Marangoni, Tanja Miletic, Jenifer Rubio-Magnieto, John Mohanraj, Heinz Amenitsch, Dario Pasini, Nikos Liaros, Stelios Couris, Nicola Armaroli, Mathieu Surin, Davide Bonifazi
DOI: 10.1021/jacs.5b02448
J. Am. Chem. Soc.
1 Jul 2015
Abstract
The self-assembly and self-organization behavior of uracil-conjugated enantiopure (R)- or (S)-1,1-binaphthyl-2,2-diol (BINOL) and a hydrophobic oligo(p-phenylene ethynylene) (OPE) chromophore exposing 2,6-di(acetylamino)pyridine termini are reported. Systematic spectroscopic (UVvis, CD, fluorescence, NMR, and SAXS) and microscopic studies (TEM and AFM) showed that BINOL and OPE compounds undergo triple H-bonding recognition, generating different organic nanostructures in solution. Depending on the solvophobic properties of the liquid media (toluene, CHCl3, CHCl3/CHX, and CHX/THF), spherical, rod-like, fibrous, and helical morphologies were obtained, with the latter being the only nanostructures expressing chirality at the microscopic level. SAXS analysis combined with molecular modeling simulations showed that the helical superstructures are composed of dimeric double-cable tape-like structures that, in turn, are supercoiled at the microscale. This behavior is interpreted as a consequence of an interplay among the degree of association of the H-bonded recognition, the vapor pressure of the solvent, and the solvophobic/solvophilic character of the supramolecular adducts in the different solutions under static and dynamic conditions, namely solvent evaporation conditions at room temperature.
Magnetically Active Carbon Nanotubes at Work
Antoine Stopin, Florent Pineux, Riccardo Marega, Davide Bonifazi
Journal
Chem.-Eur. J.
Date
06/2015
Chem.-Eur. J.
06/2015
Magnetically Active Carbon Nanotubes at Work
Antoine Stopin, Florent Pineux, Riccardo Marega, Davide Bonifazi
Chem.-Eur. J.
22 Jun 2015
Abstract
Endohedral and exohedral assembly of magnetic nanoparticles (MNPs) and carbon nanotubes (CNTs) recently gave birth to a large body of new hybrid nanomaterials (MNPs-CNTs) featuring properties that are otherwise not in reach with only the graphitic or metallic cores themselves. These materials feature enhanced magnetically guided motions (rotation and translation), magnetic saturation and coercivity, large surface area, and thermal stability. By guiding the reader through the most significant examples in this Concept paper, we describe how researchers in the field engineered and exploited the synergistic combination of these two types of nanoparticles in a large variety of current and potential applications, such as magnetic fluid hyperthermia therapeutics and in magnetic resonance imaging to name a few.
Orthogonal Insertion of Lanthanide and Transition-Metal Atoms in Metal-Organic Networks on Surfaces
Jose I. Urgel, David Ecija, Willi Auwaerter, Daphne Stassen, Davide Bonifazi, Johannes V. Barth
Journal
Angew. Chem.-Int. Edit.
Date
05/2015
Angew. Chem.-Int. Edit.
05/2015
Orthogonal Insertion of Lanthanide and Transition-Metal Atoms in Metal-Organic Networks on Surfaces
Jose I. Urgel, David Ecija, Willi Auwaerter, Daphne Stassen, Davide Bonifazi, Johannes V. Barth
Angew. Chem.-Int. Edit.
18 May 2015
Abstract
The orthogonal coordinative properties of tetrapyrrole macrocycles and nitrile ligands have been used in a multistep procedure towards interfacial d-f hetero-bimetallic nanoarchitectures based on a free-base porphyrin derivative functionalized with meso-cyanobiphenylene substituents. Molecular-level scanning tunneling microscopy studies reveal that the porphyrin module alone self-assembles on Ag(111) in a close-packed layer with a square unit cell. Upon co-deposition of Gd atoms, a square-planar motif is formed that reflects the fourfold coordination of CN ligands to the rare-earth centers. The resulting nanoporous network morphology is retained following exposure to a beam of Co atoms, which induces selective porphyrin metalation and ultimately yields a gridlike 2D metallosupramolecular architecture.
Tailoring melanins for bioelectronics: polycysteinyldopamine as an ion conducting redox-responsive polydopamine variant for pro-oxidant thin films
Nicola Fyodor Della Vecchia, Riccardo Marega, Marianna Ambrico, Mariagrazia Iacomino, Raffaella Micillo, Alessandra Napolitano, Davide Bonifazi, Marco dIschia
DOI: 10.1039/c5tc00672d
Journal
J. Mater. Chem. C
Date
05/2015
J. Mater. Chem. C
05/2015
Tailoring melanins for bioelectronics: polycysteinyldopamine as an ion conducting redox-responsive polydopamine variant for pro-oxidant thin films
Nicola Fyodor Della Vecchia, Riccardo Marega, Marianna Ambrico, Mariagrazia Iacomino, Raffaella Micillo, Alessandra Napolitano, Davide Bonifazi, Marco dIschia
DOI: 10.1039/c5tc00672d
J. Mater. Chem. C
8 May 2015
Abstract
Polycysteinyldopamine (pCDA), a red hair-inspired polydopamine-like polymer with ionic conductor behavior, can produce smooth and highly adhesive thin films and coatings on quartz, glass and other surfaces, and is shown to markedly accelerate the autoxidation of glutathione at physiological pH via an efficient redox exchange process.
Rational Synthesis of AB-Type N-Substituted Core-Functionalized Naphthalene Diimides (cNDIs)
Andrey A. Berezin, Andrea Sciutto, Nicola Demitri, Davide Bonifazi
Journal
Org. Lett.
Date
04/2015
Org. Lett.
04/2015
Rational Synthesis of AB-Type N-Substituted Core-Functionalized Naphthalene Diimides (cNDIs)
Andrey A. Berezin, Andrea Sciutto, Nicola Demitri, Davide Bonifazi
Org. Lett.
17 Apr 2015
Abstract
Acid-mediated transformation of tetraethyl 2,6-diethoxynaphthalene-1,4,5,8-tetracarboxylate selectively affords the core-substituted naphthalene-anhydride-ester (cNAE) in quantitative yield. This anhydride can be selectively converted into hetero-N-substituted core-functionalized naphthalene diimides (cNDIs) through sequential condensation reactions in the presence of the precursor amine with very high isolated yields over four steps. The approach can be applied to prepare a large variety of heterocyclic, aromatic, and aliphatic heterodiimides.
Controlling Coordination Reactions and Assembly on a Cu(111) Supported Boron Nitride Monolayer
Jose I. Urgel, Martin Schwarz, Manuela Garnica, Daphne Stassen, Davide Bonifazi, David Ecija, Johannes V. Barth, Willi Auwaerter
DOI: 10.1021/ja511611r
Journal
J. Am. Chem. Soc.
Date
02/2015
J. Am. Chem. Soc.
02/2015
Controlling Coordination Reactions and Assembly on a Cu(111) Supported Boron Nitride Monolayer
Jose I. Urgel, Martin Schwarz, Manuela Garnica, Daphne Stassen, Davide Bonifazi, David Ecija, Johannes V. Barth, Willi Auwaerter
DOI: 10.1021/ja511611r
J. Am. Chem. Soc.
25 Feb 2015
Abstract
We report the formation of a metal-organic network on a BN/Cu(111) template by codeposition of carbonitrile-functionalized porphyrin derivatives (2H-TPCN) with Co atoms in an ultrahigh vacuum environment. The resulting metallo-supramolecular structure explored by scanning tunneling microscopy and spectroscopy features a distinct 4-fold coordination motif. Furthermore, we demonstrate an in situ metalation of the tetrapyrrole macrocycles with deposited Co atoms yielding Co-TPCN directly on the BN sheet. Our results provide perspectives for the formation of coordination networks on BN and related systems featuring structural, electronic, and magnetic properties unachievable on metallic supports.
Chiral nanostructuring of multivalent macrocycles in solution and on surfaces
Marco Caricato, Arnaud Delforge, Davide Bonifazi, Daniele Dondi, Andrea Mazzanti, Dario Pasini
DOI: 10.1039/c4ob02643h
Journal
Org. Biomol. Chem.
Date
01/2015
Org. Biomol. Chem.
01/2015
Chiral nanostructuring of multivalent macrocycles in solution and on surfaces
Marco Caricato, Arnaud Delforge, Davide Bonifazi, Daniele Dondi, Andrea Mazzanti, Dario Pasini
DOI: 10.1039/c4ob02643h
Org. Biomol. Chem.
20 Jan 2015
Abstract
We describe the design and synthesis of a novel functionality-rich, homochiral macrocycle, possessing the overall molecular D-2 symmetry, in which multivalency is introduced into the covalent framework by means of four suitably positioned pyridine moieties. The macrocycle synthesis is carried out with functionalized, enantiopure 1,1-binaphthyl synthons as the source of chirality by means of a room temperature esterification reaction as the cyclization procedure. Upon addition of Pd2+, coordination of the pyridine moieties occurs both intra and intermolecularly, to afford chiral ordered mono and dimeric macrocycles or multimeric aggregates depending on the solvents and conditions used. The metal binding event takes place in combination with a significant macrocyclic conformational rearrangement detected by circular dichroism spectroscopy. When in combination with a third component (C-60), the macrocycle-Pd2+ hybrid undergoes surface-confined nanostructuring into chiral nanofibres.
Versatile Bisethynyl[60]fulleropyrrolidine Scaffolds for Mimicking Artificial Light-Harvesting Photoreaction Centers
Adrian Kremer, Emerance Bietlot, Alberto Zanelli, Joanna M. Malicka, Nicola Armaroli, Davide Bonifazi
Journal
Chem.-Eur. J.
Date
01/2015
Chem.-Eur. J.
01/2015
Versatile Bisethynyl[60]fulleropyrrolidine Scaffolds for Mimicking Artificial Light-Harvesting Photoreaction Centers
Adrian Kremer, Emerance Bietlot, Alberto Zanelli, Joanna M. Malicka, Nicola Armaroli, Davide Bonifazi
Chem.-Eur. J.
12 Jan 2015
Abstract
Fullerene-based tetrads, triads, and dyads are presented in which [60]fulleropyrrolidine synthons are linked to an oligo(p-phenyleneethynylene) antenna at the nitrogen atom and to electron-donor phenothiazine (PTZ) and/or ferrocene (Fc) moieties at the carbon of the pyrrolidine cycle through an acetylene spacer. Cyclic voltammetry and UV/ Vis absorption spectra evidence negligible ground-state electronic interactions among the subunits. By contrast, strong excited-state interactions are detected upon selective light irradiation of the antenna (UV) or of the fullerene scaffold (Vis). When only PTZ is present as electron donor, photoinduced electron transfer to the fullerene unit is unambiguously detected in benzonitrile, but this is not the case when Fc is part of the multicomponent system. These results suggest that Fc is a formidable energy transfer quencher and caution should be used in choosing it as electron donor to promote efficient charge separation in multicomponent arrays.
2014
Title
Anomalous Coarsening Driven by Reversible Charge Transfer at Metal-Organic Interfaces
Ada Della Pia, Massimo Riello, Andrea Floris, Daphne Stassen, Tim S. Jones, Davide Bonifazi, Alessandro De Vita, Giovanni Costantini
DOI: 10.1021/nn505063w
Journal
ACS Nano
Date
12/2014
ACS Nano
12/2014
Anomalous Coarsening Driven by Reversible Charge Transfer at Metal-Organic Interfaces
Ada Della Pia, Massimo Riello, Andrea Floris, Daphne Stassen, Tim S. Jones, Davide Bonifazi, Alessandro De Vita, Giovanni Costantini
DOI: 10.1021/nn505063w
ACS Nano
1 Dec 2014
Abstract
The unique electronic properties and functional tunability of polycyclic aromatic hydrocarbons have recently fostered high hopes for their use in flexible, green, portable, and cheap technologies. Most applications require the deposition of thin molecular films onto conductive electrodes. The growth of the first few molecular layers represents a crucial step in the device fabrication since it determines the structure of the molecular film and the energy level alignment of the metal-organic interface. Here, we explore the formation of this interface by analyzing the interplay between reversible molecule-substrate charge transfer, yielding intermolecular repulsion, and van der Waals attractions in driving the molecular assembly. Using a series of ad hoc designed molecules to balance the two effects, we combine scanning tunnelling microscopy with atomistic simulations to study the self-assembly behavior. Our systematic analysis identifies a growth mode characterized by anomalous coarsening that we anticipate to occur in a wide class of metal-organic interfaces and which should thus be considered as integral part of the self-assembly process when depositing a molecule on a conducting surface.
89Zr-labeled anti-endoglin antibody-targeted gold nanoparticles for imaging cancer: implications for future cancer therapy
Linda Karmani, Virginie Bouchat, Caroline Bouzin, Philippe Leveque, Daniel Labar, Anne Bol, Gladys Deumer, Riccardo Marega, Davide Bonifazi, Vincent Haufroid, Carine Michiels, Vincent Gregoire, Olivier Feron, Stephane Lucas, Thierry Vander Borght, Bernard Gallez
DOI: 10.2217/NNM.13.185
Journal
Nanomedicine
Date
10/2014
Nanomedicine
10/2014
89Zr-labeled anti-endoglin antibody-targeted gold nanoparticles for imaging cancer: implications for future cancer therapy
Linda Karmani, Virginie Bouchat, Caroline Bouzin, Philippe Leveque, Daniel Labar, Anne Bol, Gladys Deumer, Riccardo Marega, Davide Bonifazi, Vincent Haufroid, Carine Michiels, Vincent Gregoire, Olivier Feron, Stephane Lucas, Thierry Vander Borght, Bernard Gallez
DOI: 10.2217/NNM.13.185
Nanomedicine
24 Oct 2014
Abstract
Aims: Antibody-labeled gold nanoparticles represent an attractive tool for cancer imaging and therapy. In this study, the anti-CD105 antibody was conjugated with gold nanoparticles (AuNPs) for the first time. The antibody biodistribution in mice before and after conjugation to AuNPs was studied, with a focus on tumor targeting. Materials & methods: Antibodies were radiolabeled with 89Zr before conjugation to AuNPs (5 nm). Immunonanoconjugates were characterized in vitro in terms of size, stability in plasma and binding to the target. Quantitative PET imaging and ICP-MS ana-lysis assessed in vivo distribution and specific tumor targeting of tracers. Results: The tumor uptake of immunoconjugates was preserved up to 24h after injection, with high tumor contrast and selective tumor targeting. No major tracer accumulation was observed over time in nonspecific organs. ICP-MS ana-lysis confirmed the antibody specificity after nanoparticle conjugation. Conclusion: The anti-CD105 antibody conjugation to AuNPs did not greatly affect CD105-dependent tumor uptake and the efficacy of tumor targeting for cancer detection.
Hierarchical Self-Assembly of Supramolecular Hydrophobic Metallacycles into Ordered Nanostructures
Jing Zhang, Riccardo Marega, Li-Jun Chen, Nai-Wei Wu, David C. Muddiman, Davide Bonifazi, Hai-Bo Yang
Journal
Chem.-Asian J.
Date
10/2014
Chem.-Asian J.
10/2014
Hierarchical Self-Assembly of Supramolecular Hydrophobic Metallacycles into Ordered Nanostructures
Jing Zhang, Riccardo Marega, Li-Jun Chen, Nai-Wei Wu, David C. Muddiman, Davide Bonifazi, Hai-Bo Yang
Chem.-Asian J.
1 Oct 2014
Abstract
We describe herein the hierarchical self-assembly of discrete supramolecular metallacycles into ordered fibers or spherical particles through multiple noncovalent interactions. A new series of well-defined metallacycles decorated with long alkyl chains were obtained through metal-ligand interactions, which were capable of aggregating into ordered fibroid or spherical nanostructures on the surface, mostly driven by hydrophobic interactions. In-depth studies indicated that the morphology diversity was originated from the structural information encoded in the metallacycles, including the number of alkyl chains and their spatial orientation. Interestingly, the morphology of the metallacycle aggregates could be tuned by changing the solvent polarity. These findings are of special significance since they provide a simple yet highly controllable approach to prepare ordered and tunable nanostructures from small building blocks by means of hierarchical self-assembly.
Self-Assembly of Decoupled Borazines on Metal Surfaces: The Role of the Peripheral Groups
Nataliya Kalashnyk, Praveen Ganesh Nagaswaran, Simon Kervyn, Massimo Riello, Ben Moreton, Tim S. Jones, Alessandro De Vita, Davide Bonifazi, Giovanni Costantini
Journal
Chem.-Eur. J.
Date
09/2014
Chem.-Eur. J.
09/2014
Self-Assembly of Decoupled Borazines on Metal Surfaces: The Role of the Peripheral Groups
Nataliya Kalashnyk, Praveen Ganesh Nagaswaran, Simon Kervyn, Massimo Riello, Ben Moreton, Tim S. Jones, Alessandro De Vita, Davide Bonifazi, Giovanni Costantini
Chem.-Eur. J.
8 Sep 2014
Abstract
Two borazine derivatives have been synthesised to investigate their self-assembly behaviour on Au(111) and Cu(111) surfaces by scanning tunnelling microscopy (STM) and theoretical simulations. Both borazines form extended 2D networks upon adsorption on both substrates at room temperature. Whereas the more compact triphenyl borazine 1 arranges into close-packed ordered molecular islands with an extremely low density of defects on both substrates, the tris(phenyl-4-phenylethynyl) derivative 2 assembles into porous molecular networks due to its longer lateral substituents. For both species, the steric hindrance between the phenyl and mesityl substituents results in an effective decoupling of the central borazine core from the surface. For borazine 1, this is enough to weaken the molecule-substrate interaction, so that the assemblies are only driven by attractive van der Waals intermolecular forces. For the longer and more flexible borazine 2, a stronger molecule-substrate interaction becomes possible through its peripheral substituents on the more reactive copper surface.
2,5-Diamide-Substituted Five-Membered Heterocycles: Challenging Molecular Synthons
Chiara Fabbro, Simone Armani, Laure-Elie Carloni, Federica De Leo, Johan Wouters, Davide Bonifazi
Journal
Eur. J. Org. Chem.
Date
09/2014
Eur. J. Org. Chem.
09/2014
2,5-Diamide-Substituted Five-Membered Heterocycles: Challenging Molecular Synthons
Chiara Fabbro, Simone Armani, Laure-Elie Carloni, Federica De Leo, Johan Wouters, Davide Bonifazi
Eur. J. Org. Chem.
1 Sep 2014
Abstract
We describe synthetic routes for preparing previously unknown 2,5-diamide-substituted five-membered heterocycles based on the thiophene, pyrrole, and furan ring systems by exploiting Curtius-like rearrangement reactions. Conformation analysis of the 2,5-diamidothiophene derivatives identified a closed conformation, in which the two carbonyl O atoms are in close contact with the thiophene S atom.
Coverage-Dependent Disorder-to-Order Phase Transformation of a Uracil Derivative on Ag(111)
Mihaela Enache, Laura Maggini, Anna Llanes-Pallas, Thomas A. Jung, Davide Bonifazi, Meike Stohr
DOI: 10.1021/jp503188m
Journal
J. Phys. Chem. C
Date
07/2014
J. Phys. Chem. C
07/2014
Coverage-Dependent Disorder-to-Order Phase Transformation of a Uracil Derivative on Ag(111)
Mihaela Enache, Laura Maggini, Anna Llanes-Pallas, Thomas A. Jung, Davide Bonifazi, Meike Stohr
DOI: 10.1021/jp503188m
J. Phys. Chem. C
17 Jul 2014
Abstract
The self-organization of an angular bis(uracil-ethynyl) benzene derivative is investigated on Ag(111) by means of scanning tunneling microscopy (STM) under ultrahigh vacuum (UHV) conditions. It is found-starting at low submonolayer coverage that upon increasing the molecular coverage a disorder-to-order phase transformation occurs. Specifically, at low and intermediate molecular coverage a glassy phase consisting of one-dimensional (1D) chains and 2D aggregates is observed, while close to a first complete molecular layer, a well-ordered 2D close-packed phase is revealed. The main driving forces responsible for the structure formation are (i) the high self-complementarity of the uracil (U) moiety, resulting in U-U homopairs through 2-fold C=O center dot center dot center dot H-N H-bonds and (ii) the steric hindrance induced in the system by the alkyl chains. The delicate balance between the molecule molecule and the molecule substrate interactions leads to a complex phase behavior of the uracil derivative at the solid vacuum interface. On the basis of this detailed study, we present a qualitative understanding of the peculiar phase behavior of the system.
Self-Organization of Polar Porphyrinoids
Sukumaran Santhosh Babu, Davide Bonifazi
Journal
ChemPlusChem
Date
07/2014
ChemPlusChem
07/2014
Self-Organization of Polar Porphyrinoids
1 Jul 2014
Abstract
This Minireview focuses on the very recent developments in the field of porphyrin-based self-assembled nanomaterials with a particular focus on the use of tetrapyrrolic macrocycles bearing amphiphilic and ionic functionalities. As one of the most studied molecular materials, the importance of porphyrin-derived nanomaterials has attracted wide interest in many applications like catalysis, photodynamic therapy, and organic solar cells to name a few. By using key examples that have recently appeared in the literature, the discussion unfolds through two sections: amphiphilic and ionic assemblies. Although it focuses on the nanostructuring methodologies used to obtain organized materials, the discussion will also target the physical characterization of the functional materials for their implementation in prototypes.
Phenanthroline-functionalized MWCNTs as versatile platform for lanthanides complexation
Laura Maggini, Jennifer Ahrens-Jensen, Giada Lo Re, Jean-Marie Raquez, Philippe Dubois, Davide Bonifazi
Journal
Carbon
Date
04/2014
Carbon
04/2014
Phenanthroline-functionalized MWCNTs as versatile platform for lanthanides complexation
Laura Maggini, Jennifer Ahrens-Jensen, Giada Lo Re, Jean-Marie Raquez, Philippe Dubois, Davide Bonifazi
Carbon
1 Apr 2014
Abstract
A multiwalled carbon nanotube (MWCNT) scaffold was covalently functionalized with phenanthroline moieties capable to chelate tris Eu(III) complexes, such as Eu(III) tris(2-theonyl)-trifluoroacetonate ([EuL3]), yielding a brightly luminescent hybrid (MWCNTs-Phene center dot[EuL3]). The material was thoroughly characterized by means of TGA, XPS, TEM and steady-state UV-Vis absorption and emission investigations. These studies demonstrated both the integrity of the luminescent Eu(III)-based complex in the hybrid, as well. as its high loading. The versatility of the coordinating properties of phenanthroline allowed the anchoring of other lanthanides like Gd(III), producing functional hybrids with potential applicability as magnetic resonance agents. Finally, the developed hybrid revealed to be highly dispersible in biodegradable polymer matrices such as Poly(L-lactide) (PLLA), making it a promising luminophore for applications in biomaterial science. (C) 2013 Elsevier Ltd. All rights reserved.
Filling carbon nanotubes for nanobiotechnological applications
Riccardo Marega, Davide Bonifazi
DOI: 10.1039/c3nj01008b
Journal
New J. Chem.
Date
01/2014
New J. Chem.
01/2014
Filling carbon nanotubes for nanobiotechnological applications
1 Jan 2014
Abstract
With the great development in new filling methodologies for preparing endohedral carbon nanotubes, encapsulation strategies employing biomedically relevant molecular guests have emerged rapidly in recent years. All of these hybrid nanomaterials feature distinct properties and potential applications depending on both the chemical nature and spatial arrangement of the encapsulated molecular guests. In this focus article, we discuss the most significant examples in which carbon nanotube (CNTs) hybrids, filled with suitable molecular species, are used for biomedical applications. CNTs containing strongly emitting molecules hold great promises for diagnostic devices, whereas those filled with radioactive species and magnetically-active nanoparticles are attracting considerable attention for theranostic applications. Examples describing the use of the CNTs tubular cavity as an active reservoir for the controlled release of drugs are also discussed.
2013
Title
Fullerene-driven encapsulation of a luminescent Eu(III) complex in carbon nanotubes
Laura Maggini, Melinda-Emese Fuestoes, Thomas W. Chamberlain, Cristina Cebrian, Mirco Natali, Marek Pietraszkiewicz, Oksana Pietraszkiewicz, Edit Szekely, Katalin Kamaras, Luisa De Cola, Andrei N. Khlobystov, Davide Bonifazi
DOI: 10.1039/c3nr05876j
Journal
Nanoscale
Date
12/2013
Nanoscale
12/2013
Fullerene-driven encapsulation of a luminescent Eu(III) complex in carbon nanotubes
Laura Maggini, Melinda-Emese Fuestoes, Thomas W. Chamberlain, Cristina Cebrian, Mirco Natali, Marek Pietraszkiewicz, Oksana Pietraszkiewicz, Edit Szekely, Katalin Kamaras, Luisa De Cola, Andrei N. Khlobystov, Davide Bonifazi
DOI: 10.1039/c3nr05876j
Nanoscale
12 Dec 2013
Abstract
A novel CNT-based hybrid luminescent material was obtained via encapsulation of a C-60-based Eu(III) complex into single-, double-and multi-walled carbon nanotubes (SWCNTs, DWCNTs and MWCNTs, respectively). Specifically, a luminescent negatively charged Eu(III) complex, electrostatically bonded to an imidazolium-functionalized fullerene cage, was transported inside CNTs by exploiting the affinity of fullerenes for the inner surface of these carbonaceous containers. The filling was performed under supercritical CO2 (scCO(2)) conditions to facilitate the entrapment of the ion-paired assembly. Accurate elemental, spectroscopic and morphological characterization not only demonstrated the efficiency of the filling strategy, but also the occurrence of nano-ordering of the encapsulated supramolecular luminophores when SWCNTs were employed.
Supramolecular Assembly of Interfacial Nanoporous Networks with Simultaneous Expression of Metal-Organic and Organic-Bonding Motifs
Saranyan Vijayaraghavan, David Ecija, Willi Auwaerter, Sushobhan Joshi, Knud Seufert, Mateusz Drach, Damian Nieckarz, Pawel Szabelski, Claudia Aurisicchio, Davide Bonifazi, Johannes V. Barth
Journal
Chem.-Eur. J.
Date
10/2013
Chem.-Eur. J.
10/2013
Supramolecular Assembly of Interfacial Nanoporous Networks with Simultaneous Expression of Metal-Organic and Organic-Bonding Motifs
Saranyan Vijayaraghavan, David Ecija, Willi Auwaerter, Sushobhan Joshi, Knud Seufert, Mateusz Drach, Damian Nieckarz, Pawel Szabelski, Claudia Aurisicchio, Davide Bonifazi, Johannes V. Barth
Chem.-Eur. J.
11 Oct 2013
Abstract
The formation of 2D surface-confined supramolecular porous networks is scientifically and technologically appealing, notably for hosting guest species and confinement phenomena. In this study, we report a scanning tunneling microscopy (STM) study of the self-assembly of a tripod molecule specifically equipped with pyridyl functional groups to steer a simultaneous expression of lateral pyridyl-pyridyl interactions and Cu-pyridyl coordination bonds. The assembly protocols yield a new class of porous open assemblies, the formation of which is driven by multiple interactions. The tripod forms a purely porous organic network on Ag(111), phase , in which the presence of the pyridyl groups is crucial for porosity, as confirmed by molecular dynamics and Monte Carlo simulations. Additional deposition of Cu dramatically alters this scenario. For submonolayer coverage, three different porous phases coexist (i.e., , , and ). Phases and are chiral and exhibit a simultaneous expression of lateral pyridyl-pyridyl interactions and twofold Cu-pyridyl linkages, whereas phase is just stabilized by twofold Cu-pyridyl bonds. An increase in the lateral molecular coverage results in a rise in molecular pressure, which leads to the formation of a new porous phase (epsilon), only coexisting with phase and stabilized by a simultaneous expression of lateral pyridyl-pyridyl interactions and threefold Cu-pyridyl bonds. Our results will open new avenues to create complex porous networks on surfaces by exploiting components specifically designed for molecular recognition through multiple interactions.
NLO Response of Photoswitchable Azobenzene-Based Materials
Nikolaos Liaros, Stelios Couris, Laura Maggini, Federica De Leo, Fabrizio Cattaruzza, Claudia Aurisicchio, Davide Bonifazi
Journal
ChemPhysChem
Date
09/2013
ChemPhysChem
09/2013
NLO Response of Photoswitchable Azobenzene-Based Materials
Nikolaos Liaros, Stelios Couris, Laura Maggini, Federica De Leo, Fabrizio Cattaruzza, Claudia Aurisicchio, Davide Bonifazi
ChemPhysChem
16 Sep 2013
Abstract
The nonlinear optical (NLO) response of three -conjugated azobenzene (AB) derivatives was investigated under picosecond laser excitation by means of the Z-scan technique to evaluate the effect of an ethynyl-based conjugated spacer on the NLO properties of ABs. All modules possessed large third-order nonlinearity, but unexpectedly it was the less extended AB derivative that exhibited the largest NLO response. This finding has been confirmed by means of DFT calculations and was attributed to a higher cis/trans ratio of the particular AB derivative in its investigated photoequilibrated state. Furthermore, the influence of the amount of cis isomer on the third-order nonlinear susceptibility [((3))] of the less extended AB derivative has been thoroughly investigated. Specifically, modulation of the NLO response has been successfully achieved by tuning the isomeric composition of the investigated photostationary state. These results highlighted the cis-dependent increase of the NLO response to support the general idea that such compounds can be used for multistep switching NLO materials.
Structural and Dynamic Properties of Monoclonal Antibodies Immobilized on CNTs: A Computational Study
Federica De Leo, Jacopo Sgrignani, Davide Bonifazi, Alessandra Magistrato
Journal
Chem.-Eur. J.
Date
09/2013
Chem.-Eur. J.
09/2013
Structural and Dynamic Properties of Monoclonal Antibodies Immobilized on CNTs: A Computational Study
Federica De Leo, Jacopo Sgrignani, Davide Bonifazi, Alessandra Magistrato
Chem.-Eur. J.
9 Sep 2013
Abstract
Due to the widespread application of carbon nanotube (CNT)-based materials in nanomedicine, it is nowadays of paramount importance to unravel at the atomistic level of detail the structural properties of such bioconjugates in order to rationalize and predict the effect exerted by the graphitic framework on the bio-active counterpart. In this paper, we report for the first time all-atom explicit solvent molecular dynamics (MD) simulations investigating the structural and dynamic properties of a noncovalent bioconjugate in which the monoclonal Cetuximab antibody (Ctx) is adsorbed on a CNT surface. Upon selection of the three most representative adsorption modes as obtained by docking studies, force-field MD and DFT simulations unambiguously showed that hydrophobic interactions mainly govern the adsorption of the protein on the graphitic surface. Two main adsorption poses have been predicted: a pose-fab (p-fab) and pose-fc (p-fc) (fab = fragment antigen binding region; fc = fragment crystallizable region), the former being favored with small-diameter tubes (40 angstrom). In all the predicted poses, the secondary structure of Ctx is largely unaffected by the presence of the graphitic surface and, consistently with previous literature studies, our simulations reveal that positively charged amino acidic residues, such as Lys and Arg, predominantly contribute to the stabilization of the CNTCtx complex acting like surfactants. The predicted structural models are consistent with the experimental data, for which the immobilization of the antibody on CNTs does not disrupt the structural and recognition properties of the Ctx, consequently supporting the reliability of the used bioconjugation strategy for engineering stable and responsive hybrid nanomaterials for therapeutic applications. Moreover, a remarkable structural similarity of Ctx with antibodies of different isotypes suggests that in principle the CNT framework can interact in the same manner with all antibodies currently used in clinical applications.
Antibody-functionalized nanoparticles for imaging cancer: influence of conjugation to gold nanoparticles on the biodistribution of 89Zr-labeled cetuximab in mice
Linda Karmani, Daniel Labar, Vanessa Valembois, Virginie Bouchat, Praveen Ganesh Nagaswaran, Anne Bol, Jacques Gillart, Philippe Leveque, Caroline Bouzin, Davide Bonifazi, Carine Michiels, Olivier Feron, Vincent Gregoire, Stephane Lucas, Thierry Vander Borght, Bernard Gallez
DOI: 10.1002/cmmi.1539
Journal
Contrast Media Mol. Imaging
Date
09/2013
Contrast Media Mol. Imaging
09/2013
Antibody-functionalized nanoparticles for imaging cancer: influence of conjugation to gold nanoparticles on the biodistribution of 89Zr-labeled cetuximab in mice
Linda Karmani, Daniel Labar, Vanessa Valembois, Virginie Bouchat, Praveen Ganesh Nagaswaran, Anne Bol, Jacques Gillart, Philippe Leveque, Caroline Bouzin, Davide Bonifazi, Carine Michiels, Olivier Feron, Vincent Gregoire, Stephane Lucas, Thierry Vander Borght, Bernard Gallez
DOI: 10.1002/cmmi.1539
Contrast Media Mol. Imaging
1 Sep 2013
Abstract
Antibody-labeled gold nanoparticles represent a promising novel tool regarding cancer imaging and therapy. Nevertheless, the characterization of biodistribution of such immunonanocarriers has been poorly documented. In this study, the biodistribution of 89Zr-labeled cetuximab before and after the coupling reaction to gold nanoparticles (AuNPs) was compared and the quantitative imaging performance of 89Zr immuno-PET was evaluated. Cetuximab was functionalized with the desferal moiety and labeled with 89Zr (89Zr-Df-Bz-NCS-cetuximab). AuNPs with a mean diameter of 5 nm were synthesized according a new method developed in the laboratory, and conjugated to 89Zr-Df-Bz-NCS-cetuximab using carbodiimide chemistry (AuNPs-PPAA-cetuximab-89Zr). The two tracers were injected in A431 xenograft-bearing mice. Tumor and liver uptakes were assessed at different times after injection using quantitative PET imaging. The in vivo specificity of the binding was investigated using a saturating dose of unlabeled cetuximab. Radiolabeled cetuximab was conjugated to AuNPs with a coupling reaction yield >75%. All conjugates were stablein vitro and to a lesser extent in plasma. In vivo distribution studies revealed no significant difference in tumor uptake for cetuximab conjugated to nanoparticles up to 72 h after injection, compared with unconjugated cetuximab. Immuno-PET studies showed that AuNPs-PPAA-cetuximab-89Zr provided high tumor-to-background ratio. The liver uptake of AuNPs-PPAA-cetuximab-89Zr was higher, compared with 89Zr-Df-Bz-NCS-cetuximab. In vivo blocking experiments demonstrated selective tumor targeting after coupling reaction. This study showed that the conjugation of AuNPs to cetuximab did not affect its tumor accumulation and that the efficacy of EGFR-targeted nanoparticles was unaltered. The 89Zr-labeled cetuximab-targeted gold nanoparticles could be a valuable tool for theranostic purposes. Copyright (c) 2013 John Wiley & Sons, Ltd.
Magic Surface Clustering of Borazines Driven by Repulsive Intermolecular Forces
Simon Kervyn, Nataliya Kalashnyk, Massimo Riello, Ben Moreton, Jonathan Tasseroul, Johan Wouters, Tim S. Jones, Alessandro De Vita, Giovanni Costantini, Davide Bonifazi
Journal
Angew. Chem.-Int. Edit.
Date
07/2013
Angew. Chem.-Int. Edit.
07/2013
Magic Surface Clustering of Borazines Driven by Repulsive Intermolecular Forces
Simon Kervyn, Nataliya Kalashnyk, Massimo Riello, Ben Moreton, Jonathan Tasseroul, Johan Wouters, Tim S. Jones, Alessandro De Vita, Giovanni Costantini, Davide Bonifazi
Angew. Chem.-Int. Edit.
15 Jul 2013
Abstract
Its a kind of magic: Hydroxy pentaaryl borazine molecules self-assemble into small clusters (see structure) on Cu(111) surfaces, whereas with symmetric hexaaryl borazine molecules large islands are obtained. Simulations indicate that the observed “magic” cluster sizes result from long-range repulsive Coulomb forces arising from the deprotonation of the BOH groups of the hydroxy pentaaryl borazine.
Functionalized Fe-Filled Multiwalled Carbon Nanotubes as Multifunctional Scaffolds for Magnetization of Cancer Cells
Riccardo Marega, Federica De Leo, Florent Pineux, Jacopo Sgrignani, Alessandra Magistrato, Anil Damodar Naik, Yann Garcia, Lionel Flamant, Carine Michiels, Davide Bonifazi
Journal
Adv. Funct. Mater.
Date
07/2013
Adv. Funct. Mater.
07/2013
Functionalized Fe-Filled Multiwalled Carbon Nanotubes as Multifunctional Scaffolds for Magnetization of Cancer Cells
Riccardo Marega, Federica De Leo, Florent Pineux, Jacopo Sgrignani, Alessandra Magistrato, Anil Damodar Naik, Yann Garcia, Lionel Flamant, Carine Michiels, Davide Bonifazi
Adv. Funct. Mater.
5 Jul 2013
Abstract
With the aim to design addressable magnetically-active carbon nanotubes (CNTs) for cancer treatment, the use of Fe-filled CNTs (Fe@MWCNTs) as multifunctional scaffolds is reported for exohedrally anchoring a monoclonal antibody (mAb) known to bind a plasma membrane receptor over-expressed in several cancer cells (EGFR). Comprehensive microscopic (transmission electron microscopy, atomic force microscopy, and scanning electron microscopy) and spectroscopic (Raman, Fe-57 Mossbauer, energy dispersive spectroscopy, X-ray photoelectron spectroscopy (XPS), X-ray diffraction) characterizations reveal the efficient confinement of magnetically-active Fe phases (-Fe and Fe3C), while compositional evaluations through XPS, thermogravimetric analysis and gel electrophoresis confirm that mAb immobilization onto Fe@MWCNTs occurs. Enzyme-linked immunosorbent assay (ELISA), confocal microscopy imaging and western blotting confirm the targeting action toward EGFR-overexpressing cell lines (EGFR+). In vitro magnetic filtration experiments demonstrate that a selective removal of EGFR+ cells from a mixed population of healthy cell lines could be obtained in very short times (approximate to 10 min). Cytotoxicity evaluations by classic cell staining procedures after application of an electromagnetic radiation inducing magnetic fluid hyperthermia (MFH), show a selective suppression of the EGFR+ cell line. Molecular dynamics and docking simulations of the hybrid mAb/Fe@MWCNTs conjugates nicely show how the presence of the CNT framework does not sterically affect the conformational properties of the two antigen binding regions, further supporting the biochemical findings.
Polymorphism, Fluorescence, and Optoelectronic Properties of a Borazine Derivative
Simon Kervyn, Oliver Fenwick, Francesco Di Stasio, Yong Sig Shin, Johan Wouters, Gianluca Accorsi, Silvio Osella, David Beljonne, Franco Cacialli, Davide Bonifazi
Journal
Chem.-Eur. J.
Date
06/2013
Chem.-Eur. J.
06/2013
Polymorphism, Fluorescence, and Optoelectronic Properties of a Borazine Derivative
Simon Kervyn, Oliver Fenwick, Francesco Di Stasio, Yong Sig Shin, Johan Wouters, Gianluca Accorsi, Silvio Osella, David Beljonne, Franco Cacialli, Davide Bonifazi
Chem.-Eur. J.
1 Jun 2013
Abstract
We have prepared a new borazine derivative that bears mesityl substituents at the boron centers and displays exceptional chemical stability. Detailed crystallographic and solid-state fluorescence characterizations revealed the existence of several polymorphs, each of which showed different emission profiles. In particular, a bathochromic shift is observed when going from the lower- to the higher-density crystal. Computational investigations of the conformational dynamics of borazine 1 in both the gas phase and in the solid state using molecular dynamics (MD) simulations showed that the conformation of the peripheral aryl groups significantly varies when going from an isolated molecule (in which the rings are able to flip over the 90 degrees barrier at RT) to the crystals (in which the rotation is locked by packing effects), thus generating specific nonsymmetric intermolecular interactions in the different polymorphs. To investigate the optoelectronic properties of these materials by fabrication and characterization of light-emitting diodes (LEDs) and light-emitting electrochemical cells (LECs), borazine 1 was incorporated as the active material in the emissive layer. The current and radiance versus voltage characteristics, as well as the electroluminescence spectra reported here for the first time are encouraging prospects for the engineering of future borazine-based devices.
Nano- and microstructuration of supramolecular materials driven by H-bonded uracil.2,6-diamidopyridine complexes
Tomas Marangoni, Davide Bonifazi
DOI: 10.1039/c3nr01711g
Journal
Nanoscale
Date
05/2013
Nanoscale
05/2013
Nano- and microstructuration of supramolecular materials driven by H-bonded uracil.2,6-diamidopyridine complexes
29 May 2013
Abstract
In the last few decades, multiple H-bonded arrays have been shown to be versatile tools to prepare functional supramolecular materials. Supramolecular complexes formed by uracil (Ur) and 2,6-diamidopyridine (DAP) developed by Lehn are the first examples of multiple H-bonded systems governing the formation of supramolecular polymers in solution. Although a large variety of complementary multiple H-bonded complexes has been prepared, the use of the heteromolecular Ur.DAP complex still remains very promising due to its ease of preparation and its intermediate association strength that ensures a dynamical character to the self-assembly and self-organisation processes. In this feature article, we report a detailed account on the results that our group has obtained in this field by designing and engineering a novel library of shape persistent molecular modules able to transfer their geometrical information to the final supramolecular architectures through the formation of Ur.DAP complexes both at the nanoscopic and microscopic levels.
Anisotropically Luminescent Hydrogels Containing Magnetically-Aligned MWCNTs-Eu(III) Hybrids
Laura Maggini, Mingjie Liu, Yasuhiro Ishida, Davide Bonifazi
Journal
Adv. Mater.
Date
05/2013
Adv. Mater.
05/2013
Anisotropically Luminescent Hydrogels Containing Magnetically-Aligned MWCNTs-Eu(III) Hybrids
Laura Maggini, Mingjie Liu, Yasuhiro Ishida, Davide Bonifazi
Adv. Mater.
7 May 2013
Abstract
The anisotropic emission properties of an Eu(III)-MWCNTs-based nano-composite PNIPAAm hydrogel is induced upon application of a 10 T magnetic field, the latter dictating the alignment of the carbon nanotubes. This structuration creates directional highways for light to be preferentially absorbed, giving rise to orientation-dependent light emission intensity. Thermal control of the transparency of the aqueous matrix also allowed a stimulus-induced switching of the materials emission properties.
Magnetic Poly(vinylpyridine)-Coated Carbon Nanotubes: An Efficient Supramolecular Tool for Wastewater Purification
Laura Maggini, Jean-Marie Raquez, Riccardo Marega, Jennifer Jensen Ahrens, Florent Pineux, Franck Meyer, Philippe Dubois, Davide Bonifazi
Journal
ChemSusChem
Date
02/2013
ChemSusChem
02/2013
Magnetic Poly(vinylpyridine)-Coated Carbon Nanotubes: An Efficient Supramolecular Tool for Wastewater Purification
Laura Maggini, Jean-Marie Raquez, Riccardo Marega, Jennifer Jensen Ahrens, Florent Pineux, Franck Meyer, Philippe Dubois, Davide Bonifazi
ChemSusChem
1 Feb 2013
Abstract
Herein, we report the first example of a supramolecular carbon nanotube (CNT)-based magnetic depolluting agent for divalent metal ion (M2+) removal from aqueous solutions. In particular, magnetic multi-walled carbon nanotubes (m-MWCNTs) coated with poly(vinylpyridine) (PVPy) self-aggregate in aqueous solutions that contain divalent metal ions (such as Zn2+, Cu2+ and Pb2+) to form tight insoluble bundles in which the M2+ ions remain trapped through pyridyl-M2+-pyridyl interactions. Magnetic filtration ultimately affords the efficient separation of the depolluted solution from the precipitated M2+-CNT agglomerates. Upon acid treatment, the supramolecular threads could be disassembled to afford the free CNT-polymer hybrid, thus allowing recycling of the depolluting agent. All materials and complexation/decomplexation steps were thoroughly characterised by using thermogravimetric analysis (TGA), X-ray photoelectron spectroscopy (XPS), transmission and scanning electron microscopy (TEM and SEM, respectively). The quantification of the M2+ residual concentrations in water was evaluated by using inductively coupled plasma optical emission spectroscopy (ICP-OES), which showed that, depending on the metal cation, this material can remove up to 99% of the contaminant.
2012
Title
Azobenzene-based supramolecular polymers for processing MWCNTs
Laura Maggini, Tomas Marangoni, Benoit Georges, Joanna M. Malicka, K., Andrea Minoia, Roberto Lazzaroni, Nicola Armaroli, Davide Bonifazi
DOI: 10.1039/c2nr33358a
Journal
Nanoscale
Date
11/2012
Nanoscale
11/2012
Azobenzene-based supramolecular polymers for processing MWCNTs
Laura Maggini, Tomas Marangoni, Benoit Georges, Joanna M. Malicka, K., Andrea Minoia, Roberto Lazzaroni, Nicola Armaroli, Davide Bonifazi
DOI: 10.1039/c2nr33358a
Nanoscale
2 Nov 2012
Abstract
Photothermally responsive supramolecular polymers containing azobenzene units have been synthesised and employed as dispersants for multi-walled carbon nanotubes (MWCNTs) in organic solvents. Upon triggering the trans-cis isomerisation of the supramolecular polymer intermolecular interactions between MWCNTs and the polymer are established, reversibly affecting the suspensions of the MWCNTs, either favouring it (by heating, i.e. cis -> trans isomerisation) or inducing the CNTs precipitation (upon irradiation, trans -> cis isomerisation). Taking advantage of the chromophoric properties of the molecular subunits, the solubilisation/precipitation processes have been monitored by UV-Vis absorption spectroscopy. The structural properties of the resulting MWCNT-polymer hybrid materials have been thoroughly investigated via thermogravimetric analysis (TGA), X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM) and atomic force microscopy (AFM) and modelled with molecular dynamics simulations.
Nanostructured Cocrystals of a Borazine with [60]Fullerene
Simon Kervyn, Takashi Nakanishi, Junko Aimi, Akinori Saeki, Shu Seki, Benoit Champagne, Davide Bonifazi
DOI: 10.1246/cl.2012.1210
Journal
Chem. Lett.
Date
10/2012
Chem. Lett.
10/2012
Nanostructured Cocrystals of a Borazine with [60]Fullerene
Simon Kervyn, Takashi Nakanishi, Junko Aimi, Akinori Saeki, Shu Seki, Benoit Champagne, Davide Bonifazi
DOI: 10.1246/cl.2012.1210
Chem. Lett.
5 Oct 2012
Abstract
In this paper, the preparation and photophysical characterization of nanostructured cocrystals composed of B-trimesityl-N-triphenylborazine (1) and C-60 are reported. Preliminary results in the solid state show that the borazine-centered fluorescence is quenched in the cocrystal, displaying strong interchromophoric interaction in the solid state, as also shown by photoconductivity measurements.
CNTs in Optoelectronic Devices: New Structural and Photophysical Insights on Porphyrin-DWCNTs Hybrid Materials
Claudia Aurisicchio, Riccardo Marega, Valentina Corvaglia, John Mohanraj, Romain Delamare, Dana Alina Vlad, Cristian Kusko, Constantin Augustin Dutu, Andrea Minoia, Gaelle Deshayes, Olivier Coulembier, Sorin Melinte, Philippe Dubois, Roberto Lazzaroni, Nicola Armaroli, Davide Bonifazi
Journal
Adv. Funct. Mater.
Date
08/2012
Adv. Funct. Mater.
08/2012
CNTs in Optoelectronic Devices: New Structural and Photophysical Insights on Porphyrin-DWCNTs Hybrid Materials
Claudia Aurisicchio, Riccardo Marega, Valentina Corvaglia, John Mohanraj, Romain Delamare, Dana Alina Vlad, Cristian Kusko, Constantin Augustin Dutu, Andrea Minoia, Gaelle Deshayes, Olivier Coulembier, Sorin Melinte, Philippe Dubois, Roberto Lazzaroni, Nicola Armaroli, Davide Bonifazi
Adv. Funct. Mater.
7 Aug 2012
Abstract
The preparation and physical characterization of diverse porphyrin-derived double-walled carbon nanotubes (DWCNTs) conjugates are described. A porphyrin molecule is covalently linked and physically adsorbed to COOH-derived DWCNTs. The photophysical properties of all porphyrin-CNTs derivatives are studied in solution and in polymeric matrices. Definitive experimental evidence for photoinduced electron and/or energy transfer processes involving the porphyrin chromophores and the CNT wall is not obtained, but solid-state UV-vis absorption profiles display electronic transitions fingerprinting J- and H- type aggregates, where porphyrin molecules intermolecularly interact head-to-tail and face-to-face, respectively. In parallel, molecular modeling based on force-field simulations is performed to understand the structure of the porphyrin-CNTs interface and the nature of the interactions between the porphyrins and the DWCNTs. Finally, multilayered-type devices are fabricated with the aim of investigating the interaction of the porphyrin-derived DWCNTs with poly(3-hexylthiophene)-pyrene matrices containing small amounts of 1-[3-(methoxycarbonyl)propyl]-1-phenyl-[6.6]C61.
Antibody-functionalized polymer-coated gold nanoparticles targeting cancer cells: an in vitro and in vivo study
Riccardo Marega, Linda Karmani, Lionel Flamant, Praveen Ganesh Nageswaran, Vanessa Valembois, Bernard Masereel, Olivier Feron, Thierry Vander Borght, Stephane Lucas, Carine Michiels, Bernard Gallez, Davide Bonifazi
DOI: 10.1039/c2jm33482h
Journal
J. Mater. Chem.
Date
08/2012
J. Mater. Chem.
08/2012
Antibody-functionalized polymer-coated gold nanoparticles targeting cancer cells: an in vitro and in vivo study
Riccardo Marega, Linda Karmani, Lionel Flamant, Praveen Ganesh Nageswaran, Vanessa Valembois, Bernard Masereel, Olivier Feron, Thierry Vander Borght, Stephane Lucas, Carine Michiels, Bernard Gallez, Davide Bonifazi
DOI: 10.1039/c2jm33482h
J. Mater. Chem.
2 Aug 2012
Abstract
Gold nanoparticles (similar to 5 nm) coated with plasma-polymerized allylamine were produced through plasma vapor deposition and bioconjugated with a monoclonal antibody targeting the epidermal growth factor receptor. The resulting nanoconjugates displayed an antibody loading of about 1.7 nmol mg(-1) and efficiently target epidermal growth factor receptor overexpressing cell lines, as ascertained by ELISA and Western blot assays. The in vitro targeting properties were also confirmed in vivo, where a similar biodistribution profile of what was experienced for the unconjugated antibody was observed. Thanks to the possibility of doping the gold nanoparticles with radionuclides during plasma vapor deposition, the proposed functionalization strategy represents a very suitable platform for the in vivo cancer targeting with nanosized multifunctional particles.
Hierarchised luminescent organic architectures: design, synthesis, self-assembly, self-organisation and functions
Laura Maggini, Davide Bonifazi
DOI: 10.1039/c1cs15031f
Journal
Chem. Soc. Rev.
Date
07/2012
Chem. Soc. Rev.
07/2012
Hierarchised luminescent organic architectures: design, synthesis, self-assembly, self-organisation and functions
12 Jul 2012
Abstract
This critical review aims at highlighting the prevailing supramolecular approaches employed nowadays in the preparation of luminescent hierarchised materials. Specifically, it has the ambition to illustrate how progresses in the control of the supramolecular interaction toolbox ultimately led to the development of spectacular luminescent nano-and micro-architectures, through a combination of molecular self-assembly and self-organisation processes involving organic pi-conjugated molecules. The reader will be guided through a systematic exploration of the most common avenues to prepare and characterise luminescent self-assembled/self-organised materials embedded into one-, two- or three-dimensional networks, accompanied by a critical discussion of their main advantages and limitations. Key representative examples of this research field will be thoroughly described, with a particular focus on those systems displaying potential on the device application scene. Particular attention will be devoted to the design and synthetic approaches aimed at the preparation of the primary pi-conjugated molecular modules, the chemical, structural and electronic properties of which dramatically influence the fate and the features of the self-assembled/self-organised material (215 references).
Two-Dimensional Short-Range Disordered Crystalline Networks from Flexible Molecular Modules
David Ecija, Saranyan Vijayaraghavan, Willi Auwaerter, Sushobhan Joshi, Knud Seufert, Claudia Aurisicchio, Davide Bonifazi, Johannes V. Barth
DOI: 10.1021/nn3007948
Journal
ACS Nano
Date
05/2012
ACS Nano
05/2012
Two-Dimensional Short-Range Disordered Crystalline Networks from Flexible Molecular Modules
David Ecija, Saranyan Vijayaraghavan, Willi Auwaerter, Sushobhan Joshi, Knud Seufert, Claudia Aurisicchio, Davide Bonifazi, Johannes V. Barth
DOI: 10.1021/nn3007948
ACS Nano
1 May 2012
Abstract
Studies of complex condensed matter systems have led to the discovery of materials of unexpected spatial organization as glasses, glassy crystals, quasicrystals, and protein and virus crystals. Here, we present two-dimensional (2D) short-range disordered molecular crystalline networks, which, regarding spatial organization, can be considered as surface analogues of 3D glassy crystals. In particular, the deposition of a flexible molecular module on Cu(111) gives rise to distinct phases whose characteristics have been examined in real space by scanning tunneling microscopy: a 2D short-range distortional disordered crystalline network and a 2D short-range orientational disordered crystalline network, respectively. Both phases exhibit a random arrangement of nanopores that are stabilized by the simultaneous presence of metal-organic and pyridyl-pyridyl interactions. The 2D short-range distortional disordered crystalline network displayed intriguing flexibility, as probed by the STM tip that modifies the pore shape, a prerequisite for adaptive behavior in host-guest processes.
Luminescent Blooming of Dendronic Carbon Nanotubes through Ion-Pairing Interactions with an EuIII Complex
Laura Maggini, Francesca Maria Toma, Luigi Feruglio, Joanna M. Malicka, Tatiana Da Ros, Nicola Armaroli, Maurizio Prato, Davide Bonifazi
Journal
Chem.-Eur. J.
Date
05/2012
Chem.-Eur. J.
05/2012
Luminescent Blooming of Dendronic Carbon Nanotubes through Ion-Pairing Interactions with an EuIII Complex
Laura Maggini, Francesca Maria Toma, Luigi Feruglio, Joanna M. Malicka, Tatiana Da Ros, Nicola Armaroli, Maurizio Prato, Davide Bonifazi
Chem.-Eur. J.
1 May 2012
Abstract
A multiwalled carbon nanotube (MWCNT) scaffold was covalently functionalized with a second-generation polyamidoamine (PAMAM) dendron, presenting four terminal amino groups per grafted aryl moiety. These reactive functions were alkylated to obtain a positively charged polycationic dendron/carbon nanotube system (d-MWCNTs.Cl), which eventually underwent anion exchange reaction with a negatively charged and highly luminescent EuIII complex ([EuL4].NEt4, in which L=(2-naphtoyltrifluoroacetonate)). This process afforded the target material d-MWCNTs.[EuL4], in which MWCNTs are combined with red-emitting EuIII centers through electrostatic interactions with the dendronic branches. Characterization of the novel MWCNT materials was accomplished by means of TGA and TEM, whereas d-MWCNTs.Cl and d-MWCNTs. [EuL4] further underwent XPS, SEM and Raman analyses. These studies demonstrate the integrity of the luminescent [EuL4]- center in the luminescent hybrid, the massive load of the cationic binding sites, and the virtually complete anion-exchange into the final hybrid material. The occurrence of the ion-pairing interaction with MWCNTs was unambiguously demonstrated through DOSY NMR diffusion studies. Photophysical investigations show that MWCNTs.[EuL4] is a highly soluble and brightly luminescent red hybrid material in which MWCNTs act as photochemically inert scaffolds with negligible UV/Vis absorption, compared with the grafted Eu complex, and with no quenching activity. The high dispersibility of MWCNTs.[EuL4] in a polymer matrix makes it a promising luminophore for applications in material science.
Melting of Hydrogen Bonds in Uracil Derivatives Probed by Infrared Spectroscopy and ab Initio Molecular Dynamics
Zsolt Szekrenyes, Katalin Kamaras, Gyorgy Tarczay, Anna Llanes-Pallas, Tomas Marangoni, Maurizio Prato, Davide Bonifazi, Jonas Bjork, Felix Hanke, Mats Persson
DOI: 10.1021/jp212115h
Journal
J. Phys. Chem. B
Date
04/2012
J. Phys. Chem. B
04/2012
Melting of Hydrogen Bonds in Uracil Derivatives Probed by Infrared Spectroscopy and ab Initio Molecular Dynamics
Zsolt Szekrenyes, Katalin Kamaras, Gyorgy Tarczay, Anna Llanes-Pallas, Tomas Marangoni, Maurizio Prato, Davide Bonifazi, Jonas Bjork, Felix Hanke, Mats Persson
DOI: 10.1021/jp212115h
J. Phys. Chem. B
19 Apr 2012
Abstract
The thermal response of hydrogen bonds is a crucial aspect in the self-assembly of molecular nanostructures. To gain a detailed understanding of their response, we investigated infrared spectra of monomers and hydrogen-bonded dimers of two uracil-derivative molecules, supported by density functional theory calculations. Matrix isolation spectra of monomers, temperature dependence in the solid state, and ab initio molecular dynamics calculations give a comprehensive picture about the dimer structure and dynamics of such systems as well as a proper assignment of hydrogen-bond affected bands. The evolution of the hydrogen bond melting is followed by calculating the C=O center dot center dot center dot H-N distance distributions at different temperatures. The result this calculation yields excellent agreement with the H-bond melting temperature observed by experiment.
Multiple Hydrogen Bond Interactions in the Processing of Functionalized Multi-Walled Carbon Nanotubes
Mildred Quintana, Hassan Traboulsi, Anna Llanes-Pallas, Riccardo Marega, Davide Bonifazi, Maurizio Prato
DOI: 10.1021/nn203471t
Journal
ACS Nano
Date
01/2012
ACS Nano
01/2012
Multiple Hydrogen Bond Interactions in the Processing of Functionalized Multi-Walled Carbon Nanotubes
Mildred Quintana, Hassan Traboulsi, Anna Llanes-Pallas, Riccardo Marega, Davide Bonifazi, Maurizio Prato
DOI: 10.1021/nn203471t
ACS Nano
1 Jan 2012
Abstract
In a set of unprecedented experiments combining bottom-up and top-down approaches, we report the engineering of patterned surfaces In which functionalized MWCNTs have been selectively adsorbed on polymeric matrices as obtained by microlithographic photo-cross-linking of polystyrene polymers bearing 2,6-di(acetylamino)-4-pyridyl moieties (PS1) deposited on glass or Si. All patterned surfaces have been characterized by optical, fluorescence, and SEM imaging techniques, showing the local confinement of the CNTs materials on the polymeric microgrids. These results open new possibilities toward the controlled manipulation of CNTs on surfaces, using H-bonding self-assembly as the main driving force.
2011
Title
Electrostatically-driven assembly of MWCNTs with a europium complex
Laura Maggini, Hassan Traboulsi, K. Yoosaf, John Mohanraj, Johan Wouters, Oksana Pietraszkiewicz, Marek Pietraszkiewicz, Nicola Armaroli, Davide Bonifazi
DOI: 10.1039/c0cc02874f
Journal
Chem. Commun.
Date
11/2011
Chem. Commun.
11/2011
Electrostatically-driven assembly of MWCNTs with a europium complex
Laura Maggini, Hassan Traboulsi, K. Yoosaf, John Mohanraj, Johan Wouters, Oksana Pietraszkiewicz, Marek Pietraszkiewicz, Nicola Armaroli, Davide Bonifazi
DOI: 10.1039/c0cc02874f
Chem. Commun.
29 Nov 2011
Abstract
Luminescent carbon-based materials have been prepared by electrostatic self-assembly of negatively-charged luminescent Eu(III)-complex with imidazolium-functionalized MWCNTs.
Modular Engineering of H-Bonded Supramolecular Polymers for Reversible Functionalization of Carbon Nanotubes
Anna Llanes-Pallas, K., Hassan Traboulsi, John Mohanraj, Thomas Seldrum, Jacques Dumont, Andrea Minoia, Roberto Lazzaroni, Nicola Armaroli, Davide Bonifazi
DOI: 10.1021/ja2011516
Journal
J. Am. Chem. Soc.
Date
10/2011
J. Am. Chem. Soc.
10/2011
Modular Engineering of H-Bonded Supramolecular Polymers for Reversible Functionalization of Carbon Nanotubes
Anna Llanes-Pallas, K., Hassan Traboulsi, John Mohanraj, Thomas Seldrum, Jacques Dumont, Andrea Minoia, Roberto Lazzaroni, Nicola Armaroli, Davide Bonifazi
DOI: 10.1021/ja2011516
J. Am. Chem. Soc.
5 Oct 2011
Abstract
A H-bond-driven, noncovalent, reversible solubilization/functionalization of multiwalled carbon nanotubes (MWCNTs) in apolar organic solvents (CHCl3, CH2Cl2, and toluene) has been accomplished through a dynamic combination of self-assembly and self-organization processes leading to the formation of supramolecular polymers, which enfold around the outer wall of the MWCNTs. To this end, a library of phenylacetylene molecular scaffolds with complementary recognition sites at their extremities has been synthesized. They exhibit triple parallel H-bonds between the NH-N-NH (DAD) functions of 2,6-di(acetylamino)pyridine and the CO-NH-CO (ADA) imidic groups of uracil derivatives. These residues are placed at 180 degrees relative to each other (linear systems) or at 60 degrees/120 degrees (angular modules), in order to tune their ability of wrapping around MWCNTs. Molecular Dynamics (MD) simulations showed that the formation of the hybrid assembly MWCNT.[X.Y](n) (where X = 1a or 1b -DAD- and Y = 2, 3, or 4 -ADA-) is attributed to pi-pi and CH-pi interactions between the graphitic walls of the carbon materials and the oligophenyleneethynylene polymer backbones along with its alkyl groups, respectively. Addition of polar or protic solvents, such as DMSO or MeOH, causes the disruption of the H-bonds with partial detachment of the polymer from the CNTs, followed by precipitation. Taking advantage of the chromophoric and luminescence properties of the molecular subunits, the solubilization/precipitation processes have been monitored by UV-vis absorption and luminescence spectroscopies. All hybrid MWCNTs-polymer materials have been also structurally characterized via thermogravimetric analysis (TGA), transmission electron microscopy (TEM), atomic force microscopy (AFM), scanning tunneling microscopy (STM), and X-ray photoelectron spectroscopy (XPS).
On the route to mimic natural movements: synthesis and photophysical properties of a molecular arachnoid
Joceline Zeitouny, Abdelhalim Belbakra, Anna Llanes-Pallas, Andrea Barbieri, Nicola Armaroli, Davide Bonifazi
DOI: 10.1039/c0cc03045g
Journal
Chem. Commun.
Date
10/2011
Chem. Commun.
10/2011
On the route to mimic natural movements: synthesis and photophysical properties of a molecular arachnoid
Joceline Zeitouny, Abdelhalim Belbakra, Anna Llanes-Pallas, Andrea Barbieri, Nicola Armaroli, Davide Bonifazi
DOI: 10.1039/c0cc03045g
Chem. Commun.
4 Oct 2011
Abstract
The synthesis, photoswitchability and NIR emitting properties of a novel pi-extended pyrene derivative, peripherally decorated with four azobenzenyl-ethynyl legs, are reported.
Carbon Nanotube-Based Metal-Ion Catchers as Supramolecular Depolluting Materials
Laura Maggini, Federica De Leo, Riccardo Marega, Hajnalka-Maria Tohati, Katalin Kamaras, Davide Bonifazi
Journal
ChemSusChem
Date
09/2011
ChemSusChem
09/2011
Carbon Nanotube-Based Metal-Ion Catchers as Supramolecular Depolluting Materials
Laura Maggini, Federica De Leo, Riccardo Marega, Hajnalka-Maria Tohati, Katalin Kamaras, Davide Bonifazi
ChemSusChem
9 Sep 2011
Abstract
Herein, we report the first example of supramolecular carbon nanotube (CNT)-based ion catchers as simple and effective tools for removing divalent metal ions from organic solvents. In particular, covalently functionalized multi-walled carbon nanotubes (MWCNTs) appended with pyridyl groups self-aggregate in solution into bundles in the presence of divalent metal ions (e. g., Cd2+, Cu2+, Ni2+, Pb2+, Zn2+). Such self-aggregation behavior leads to insoluble materials that, upon treatment with weak acids, can be regenerated and reused for further complexation. All materials and complexation/decomplexation steps were thoroughly characterized by using X-ray photoelectron spectroscopy (XPS), thermogravimetric analysis (TGA), and different microscopy-based techniques, namely, transmission electron, scanning electron, and atomic force microscopy (TEM, SEM, and AFM). The supramolecular system engineered in this work is the first example of an easy and fully sustainable material with great potential applications for depolluting liquid waste from metal contamination.
[60] Fullerene-based monolayers as neuroprotective biocompatible hybrid materials
Davide Giust, Jose Luis Albasanz, Mairena Martin, Riccardo Marega, Arnaud Delforge, Davide Bonifazi
DOI: 10.1039/c1cc13971a
Journal
Chem. Commun.
Date
08/2011
Chem. Commun.
08/2011
[60] Fullerene-based monolayers as neuroprotective biocompatible hybrid materials
Davide Giust, Jose Luis Albasanz, Mairena Martin, Riccardo Marega, Arnaud Delforge, Davide Bonifazi
DOI: 10.1039/c1cc13971a
Chem. Commun.
24 Aug 2011
Abstract
Here we report on the surface immobilization of redox-active [60] fullerene derivatives and the consequent neuroprotective effects toward L-glutamate induced excitotoxicity in human derived undifferentiated neuroblastoma cells.
A Luminescent Host-Guest Hybrid between a EuIII Complex and MWCNTs
Laura Maggini, John Mohanraj, Hassan Traboulsi, Andrea Parisini, Gianluca Accorsi, Nicola Armaroli, Davide Bonifazi
Journal
Chem.-Eur. J.
Date
07/2011
Chem.-Eur. J.
07/2011
A Luminescent Host-Guest Hybrid between a EuIII Complex and MWCNTs
Laura Maggini, John Mohanraj, Hassan Traboulsi, Andrea Parisini, Gianluca Accorsi, Nicola Armaroli, Davide Bonifazi
Chem.-Eur. J.
1 Jul 2011
Abstract
Like treasure safeguarded in a chest, a tris-hexafluoro acetylacetonate EuIII complex was encapsulated inside MWCNTs to preserve its luminescent output (see figure). These structures could open the way to novel luminescent hybrid materials with potential applications in biological and materials sciences.
Engineering supramolecular photoactive nanomaterials by hydrogen-bonding interactions
K. Yoosaf, Abdelhalim Belbakra, Anna Llanes-Pallas, Davide Bonifazi, Nicola Armaroli
Journal
Pure Appl. Chem.
Date
03/2011
Pure Appl. Chem.
03/2011
Engineering supramolecular photoactive nanomaterials by hydrogen-bonding interactions
K. Yoosaf, Abdelhalim Belbakra, Anna Llanes-Pallas, Davide Bonifazi, Nicola Armaroli
Pure Appl. Chem.
14 Mar 2011
Abstract
The photophysical properties of molecules containing anthracene, pyrene, or phenyleneethynylene chromophores bearing complementary triple H-bonding terminal units, namely, 2,6-di(acetylamino) pyridine (donor-acceptor-donor, DAD) and uracyl (acceptor-donor-acceptor, ADA) have been investigated as a function of solvent polarity. For asymmetric systems, presenting only one H-bonding unit, a solvatochromic effect is found, suggesting a charge-transfer character of the lowest electronic excited state. Systematic absorption and emission studies carried out as a function of temperature show that phenyleneethynylenes having linear geometry and H-bonding functionalities at both ends undergo reversible self-aggregation in cyclohexane (CHX), leading to the formation of spherical nanoparticles, as evidenced by wide-field fluorescence microscopy (WFM), atomic force microscopy (AFM), and transmission electron microscopy (TEM). A combination of an anthracene derivative bearing only one ADA terminal functionality and a linear phenyleneethynylene derivative possessing two DAD terminal groups in CHX (2: 1 molecular ratio) leads to the formation of vesicular nanostructures. The interaction of linear phenyleneethynylenes possessing two terminal 2,6-di(acetylamino) pyridine functionalities with that bearing bis uracylic units gives origin to nanofibers, while the assembly of the former with bisuracylic units exhibiting bent geometry leads to the formation of helical nanofibers. The length of these fibers can be controlled by addition of the anthracene derivative having only one uracyl group which effectively blocks the extent of H-bonding, prompting the formation of shorter nanorods.
From Molecular to Macroscopic Engineering: Shaping Hydrogen-Bonded Organic Nanomaterials
K. Yoosaf, Anna Llanes-Pallas, Tomas Marangoni, Abdelhalim Belbakra, Riccardo Marega, Edith Botek, Benoit Champagne, Davide Bonifazi, Nicola Armaroli
Journal
Chem.-Eur. J.
Date
03/2011
Chem.-Eur. J.
03/2011
From Molecular to Macroscopic Engineering: Shaping Hydrogen-Bonded Organic Nanomaterials
K. Yoosaf, Anna Llanes-Pallas, Tomas Marangoni, Abdelhalim Belbakra, Riccardo Marega, Edith Botek, Benoit Champagne, Davide Bonifazi, Nicola Armaroli
Chem.-Eur. J.
1 Mar 2011
Abstract
The self-assembly and self-organization behavior of chromophoric acetylenic scaffolds bearing 2,6-bis(acetylamino) pyridine (1, 2) or uracyl-type (3-9) terminal groups has been investigated by photophysical and microscopic methods. Systematic absorption and luminescence studies show that 1 and 2, thanks to a combination of solvophilic/solvophobic forces and pi-pi stacking interactions, undergo self-organization in apolar solvents (i.e., cyclohexane) and form spherical nanoparticles, as evidenced by wide-field optical microscopy, TEM, and AFM analysis. For the longer molecular module, 2, a more uniform size distribution is found (80-200 nm) compared to 1 (20-1000 nm). Temperature scans in the range 283-353 K show that the self-organized nanoparticles are reversibly formed and destroyed, being stable at lower temperatures. Molecular modules 1 and 2 were then thoroughly mixed with the complementary triply hydrogen-bonding units 3-9. Depending on the specific geometrical structure of 3-9, different nanostructures are evidenced by microscopic investigations. Combination of modules 1 or 2 with 3, which bears only one terminal uracyl unit, leads to the formation of vesicular structures; instead, when 1 is combined with bis-uracyl derivative 4 or 5, a structural evolution from nanoparticles to nanowires is observed. The length of the wires obtained by mixing 1 and 4 or 1 and 5 can be controlled by addition of 3, which prompts transformation of the wires into shorter rods. The replacement of linear system 5 with the related angular modules 6 and 7 enables formation of helical nanostructures, unambiguously evidenced by AFM. Finally, thermally induced self-assembly was studied in parallel with modules 8 and 9, in which the uracyl recognition sites are protected with tert-butyloxycarbonyl (BOC) groups. This strategy allows further control of the self-assembly/self-organization process by temperature, since the BOC group is completely removed on heating. Microscopy studies show that the BOC-protected ditopic modules 8 self-assemble and self-organize with 1 into ordered linear nanostructures, whereas BOC-protected tritopic system 9 gives rise to extended domains of circular nano-objects in combination with 1.
Thermosolutal Self-Organization of Supramolecular Polymers into Nanocraters
Tomas Marangoni, Stefano A. Mezzasalma, Anna Llanes-Pallas, K. Yoosaf, Nicola Armaroli, Davide Bonifazi
DOI: 10.1021/la104276y
Journal
Langmuir
Date
02/2011
Langmuir
02/2011
Thermosolutal Self-Organization of Supramolecular Polymers into Nanocraters
Tomas Marangoni, Stefano A. Mezzasalma, Anna Llanes-Pallas, K. Yoosaf, Nicola Armaroli, Davide Bonifazi
DOI: 10.1021/la104276y
Langmuir
15 Feb 2011
Abstract
The ability of two complementary molecular modules bearing H-bonding uracilic and 2,6-(diacetylamino)pyridyl moieties to self-assemble and self-organize into submicrometer morphologies has been investigated by means of spectroscopic, thermogravimetric, and microscopic methods. Using uracilic N-3-BOC-protected modules, it has been possible to thermally trigger the self-assembly/self-organization process of the two molecular modules, inducing the formation of objects on a mica surface that exhibit crater-like morphology and a very homogeneous size distribution. Confirmation of the presence of the hydrogen-bonding-driven self-assembly/self-organization process in solution was obtained by variable-temperature (VT) steady-state UV-vis absorption and emission measurements. The variation of the geometric and spatial features of the morphologies was monitored at different T by means of atomic force microscopy (AFM) and was interpreted by a nonequilibrium diffusion model for two chemical species in solution. The formation of nanostructures turned out to be affected by the solid substrate (molecular interactions at a solid-liquid interface), by the matter-momentum transport in solution (solute diffusivity D-0 and solvent kinematic viscosity nu), and the thermally dependent cleavage reaction of the BOC functions (T-dependent differential weight loss, theta = theta(T)) in a T interval extrapolated to similar to 60 K. A scaling function, f = T(nu D-0, nu/D-0, theta), relying on the onset condition of a concentration-driven thermosolutal instability has been established to simulate the T-dependent behavior of the structural dimension (i.e., height and radius) of the self-organized nanostructures as < h > approximate to f(T) and < r > approximate to 1/f (T).
Photophysics and transient nonlinear optical response of donor-[60]fullerene hybrids
Panagiotis Aloukos, Konstantinos Iliopoulos, Stelios Couris, Dirk M. Guldi, Chloe Sooambar, Aurelio Mateo-Alonso, Praveen Ganesh Nagaswaran, Davide Bonifazi, Maurizio Prato
DOI: 10.1039/c0jm03520c
Journal
J. Mater. Chem.
Date
01/2011
J. Mater. Chem.
01/2011
Photophysics and transient nonlinear optical response of donor-[60]fullerene hybrids
Panagiotis Aloukos, Konstantinos Iliopoulos, Stelios Couris, Dirk M. Guldi, Chloe Sooambar, Aurelio Mateo-Alonso, Praveen Ganesh Nagaswaran, Davide Bonifazi, Maurizio Prato
DOI: 10.1039/c0jm03520c
J. Mater. Chem.
6 Jan 2011
Abstract
The photophysical properties of some recently synthesized donor-[60]fullerene hybrids, porphyrinyl- and ferrocenyl-[60]fullerene dyads, are studied by means of absorption, fluorescence and femtosecond transient absorption spectroscopy. The formation of charge transfer states is experimentally confirmed, while it is shown that an efficient photoinduced intra-molecular electron-transfer process takes place between the donors (i.e. porphyrin, ferrocene) and the acceptor (i.e. [60]fullerene). In addition, the transient nonlinear optical response of these [60]fullerene dyads is investigated under visible nanosecond laser excitation. Both dyads were found to exhibit significantly enhanced second hyperpolarizability (gamma) compared to that of unfunctionalized [60]fullerene, while the refractive part of their nonlinearity dominated their nonlinear optical response in contrast to what has been observed for pristine fullerene and other functionalized fullerenes. Moreover, their optical limiting action is investigated both experimentally and by means of a five-level model. The results are discussed and compared with other literature reports.
2010
Title
Supramolecular architectures of porphyrins on surfaces: The structural evolution from 1D to 2D to 3D to devices
Stefan Mohnani, Davide Bonifazi
Journal
Coord. Chem. Rev.
Date
10/2010
Coord. Chem. Rev.
10/2010
Supramolecular architectures of porphyrins on surfaces: The structural evolution from 1D to 2D to 3D to devices
1 Oct 2010
Abstract
Tetrapyrrolic macrocycles, such as porphyrins, belong to a class of distinctively multifunctional biomolecules playing a central role in fundamental natural processes such as electron transfer, oxygen transfer, and light-harvesting, and their use to mimic these biological events in nanotechnological devices would be of obvious benefit Despite the synthetic and physical achievements, a technical impediment towards the exploitation of such porphyrin-based architectures in applicative devices is that they cannot be singularly addressed in solution or at solid state, as they must be interfaced with the external world They also need to show durability and functionality under the extreme conditions that are normally used in operating practical devices. Typically, porphyrin architectures have been investigated in solution, however, the tendency in current research is to deposit functional porphyrin derivatives on the surfaces of bulk materials such as metals or semiconductors and investigate the resultant hybrid surfaces using STM. In this review, we have illustrated the trends in supramolecular nanopatterning of porphyrin derivatives at different interfaces Various strategies for the construction of nanoscale architectures at different interfaces are described along the course of the review, including sublimation under UHV conditions. adlayer formation by immersion of a surface in a liquid or deposition of a solution The discussion of assemblies on surfaces commences with a description of the very recent developments in the remarkably precisely controlled construction of discrete assemblies on surfaces under UHV conditions. Subsequently, extended 2D arrays formed in both ambient conditions at the liquid-solid interface, as well as under UHV conditions have been discussed along with the ability of certain 2D self-assemblies to accommodate guest molecules. The last section of the review deals with porphyrin assemblies featuring three-dimensional properties, with a particular focus on those systems in which the third-dimension Introduces functionality such as gas-storage, catalysis and a molecular motor (C) 2010 Elsevier B V All rights reserved.
Hierarchic Self-Assembly of Nanoporous Chiral Networks with Conformationally Flexible Porphyrins
David Ecija, Knud Seufert, Daniel Heim, Willi Auwaerter, Claudia Aurisicchio, Chiara Fabbro, Davide Bonifazi, Johannes V. Barth
DOI: 10.1021/nn1013337
Journal
ACS Nano
Date
08/2010
ACS Nano
08/2010
Hierarchic Self-Assembly of Nanoporous Chiral Networks with Conformationally Flexible Porphyrins
David Ecija, Knud Seufert, Daniel Heim, Willi Auwaerter, Claudia Aurisicchio, Chiara Fabbro, Davide Bonifazi, Johannes V. Barth
DOI: 10.1021/nn1013337
ACS Nano
1 Aug 2010
Abstract
We report the hierarchic design of homochiral 2D nanoporous networks under ultrahigh vacuum conditions on the Ag(111) surface by using a flexible porphyrin derivative as a primary unit. The conformational adaptation of the molecular module gives rise to two enantiomers upon 2D confinement, which self-assemble in enantiopure clusters made of three molecules reflecting chiral recognition, which constitute the secondary supramolecular building block mediating the formation of the tertiary complex open networks. Our results show that the creation of homochiral superstructures based on the hierarchical assembly of conformationally flexible molecular components constitutes a unique pathway toward the design of novel and functional chiral structures.
On the Aromatic Character of 1,2-Dihydro-1,2-azaborine According to Magnetic Criteria
Raphael Carion, Vincent Liegeois, Benoit Champagne, Davide Bonifazi, Stefano Pelloni, Paolo Lazzeretti
DOI: 10.1021/jz100401y
Journal
J. Phys. Chem. Lett.
Date
05/2010
J. Phys. Chem. Lett.
05/2010
On the Aromatic Character of 1,2-Dihydro-1,2-azaborine According to Magnetic Criteria
Raphael Carion, Vincent Liegeois, Benoit Champagne, Davide Bonifazi, Stefano Pelloni, Paolo Lazzeretti
DOI: 10.1021/jz100401y
J. Phys. Chem. Lett.
20 May 2010
Abstract
The behavior of benzene, 1,2-dihydro-1,2-azaborine, and borazine under a magnetic perturbation is theoretically evaluated to assess how the 1,2-dihydro-1,2-azaborine molecule displays an intermediate character between its hydrocarbon (benzene) and fully boro-nitrogen (borazine) parents. The use of tridimensional visualization tools confirms that single-value magnetic properties do not allow one to properly evaluate the whole magnetic behavior and the magnetic pi-aromaticity of these compounds, since the sigma-electron response is also modified due to the introduction of polar B-N bond(s) and has a significant influence on the various magnetic properties.
Self-Assembly of Flexible One-Dimensional Coordination Polymers on Metal Surfaces
Daniel Heim, David Ecija, Knud Seutert, Willi Auwaerter, Claudia Aurisicchio, Chiara Fabbro, Davide Bonifazi, Johannes V. Barth
DOI: 10.1021/ja1010527
Journal
J. Am. Chem. Soc.
Date
05/2010
J. Am. Chem. Soc.
05/2010
Self-Assembly of Flexible One-Dimensional Coordination Polymers on Metal Surfaces
Daniel Heim, David Ecija, Knud Seutert, Willi Auwaerter, Claudia Aurisicchio, Chiara Fabbro, Davide Bonifazi, Johannes V. Barth
DOI: 10.1021/ja1010527
J. Am. Chem. Soc.
19 May 2010
Abstract
We employed a de novo synthesized porphyrin module to construct one-dimensional (1D) Cu-coordinated polymers on Cu(111) and Ag(111) surfaces. The programmed geometry and functionality of the molecular module together with its conformational flexibility and substrate interaction yields sinuous metal-organic polymeric assemblies, based on an unusual two-fold Cu-pyridyl coordination motif. An analysis of scanning tunneling microscopy (STM) data reveals the occurrence of two enantiomers, resulting from the surface confinement that deconvolutes the module in 2D-chiral conformational isomers. The stereoisomers exhibit site-specific surface anchoring, from whence three discrete orientations are possible for each species. Their sequence and mutual arrangement determine direction and curvature of the metal-organic chains. The Cu-coordinated polymers are very similar on both Cu(111) and Ag(111), where their formation is induced by intrinsic and coevaporated adatoms, respectively, which indicates that the lateral bonding motif is predominantly independent of the substrate. In addition, molecular manipulation experiments show the collective motion of entire segments of the Cu-coordinated multi-porphyrin polymers.
Mastering nanostructured materials through H-bonding recognitions at interfaces
Stefan Mohnani, Anna Llanes-Pallas, Davide Bonifazi
Journal
Pure Appl. Chem.
Date
04/2010
Pure Appl. Chem.
04/2010
Mastering nanostructured materials through H-bonding recognitions at interfaces
1 Apr 2010
Abstract
The controlled engineering of functional architectures composed of pi-systems with unusual opto-electronic properties is currently being investigated intensively from both fundamental research and technological application viewpoints. In particular, the exploitation of the supramolecular approach for the facile construction of multidimensional architectures, featuring cavities capable of hosting functional molecules, could be used in several applications, such as nanomedicine, molecular-based memory storage devices, and sensors. This paper highlights our recent strategies to use hydrogen-bonding interactions to prepare nanostructured functional architectures via the self-assembly of organic molecular modules studied at different interfaces.
Surface-Assisted Assembly of Discrete Porphyrin-Based Cyclic Supramolecules
Daniel Heim, Knud Seufert, Willi Auwaerter, Claudia Aurisicchio, Chiara Fabbro, Davide Bonifazi, Johannes V. Barth
DOI: 10.1021/nl9029994
Journal
Nano Lett.
Date
01/2010
Nano Lett.
01/2010
Surface-Assisted Assembly of Discrete Porphyrin-Based Cyclic Supramolecules
Daniel Heim, Knud Seufert, Willi Auwaerter, Claudia Aurisicchio, Chiara Fabbro, Davide Bonifazi, Johannes V. Barth
DOI: 10.1021/nl9029994
Nano Lett.
1 Jan 2010
Abstract
We employed de novo synthesized porphyrin modules to construct discrete cyclic supramolecular architectures supported on a copper surface. The programmed geometry and functionality of the molecular modules together with their conformational flexibility and substrate interaction yields symmetric discrete assemblies, including dimers and chains as well as three- to six-membered cyclic structures. The area of the molecular cavities is extended by creating bicomponent structures combining building blocks with different symmetry.
2009
Title
Photophysical Properties of Tolan Wavelength Shifters in Solution and Embedded in Polymeric Organic Thin Films
Claudia Aurisicchio, Barbara Ventura, Davide Bonifazi, Andrea Barbieri
DOI: 10.1021/jp9053988
Journal
J. Phys. Chem. C
Date
10/2009
J. Phys. Chem. C
10/2009
Photophysical Properties of Tolan Wavelength Shifters in Solution and Embedded in Polymeric Organic Thin Films
Claudia Aurisicchio, Barbara Ventura, Davide Bonifazi, Andrea Barbieri
DOI: 10.1021/jp9053988
J. Phys. Chem. C
15 Oct 2009
Abstract
Linear organic conjugated molecules, peripherally equipped with electron-donating and electron-accepting moieties, are recognized as one of the most promising classes of nonlinear optical materials for potential application in energy conversion devices, organic electronics, optical communication, information storage, and nuclear medicine techniques. In this work, we have synthesized and photophysically characterized a series of organic molecules constituted by a 1,2-diphenyl acetylene core (tolan) bearing electronically active groups directly linked to the pi-conjugated backbone. Tuning of the absorption and emission energies has been achieved via the push-pull effect. All investigated compounds displayed very high luminescence in condensed media from intramolecular charge transfer excited states with large Stokes shifts. These features revealed to be of particular interest for the engineering of new wavelength shifters for spectral conversion of deep ultraviolet to visible light.
Tailoring Bicomponent Supramolecular Nanoporous Networks: Phase Segregation, Polymorphism, and Glasses at the Solid-Liquid Interface
Carlos-Andres Palma, Jonas Bjork, Massimo Bonini, Matthew S. Dyer, Anna Llanes-Pallas, Davide Bonifazi, Mats Persson, Paolo Samori
DOI: 10.1021/ja9032428
Journal
J. Am. Chem. Soc.
Date
09/2009
J. Am. Chem. Soc.
09/2009
Tailoring Bicomponent Supramolecular Nanoporous Networks: Phase Segregation, Polymorphism, and Glasses at the Solid-Liquid Interface
Carlos-Andres Palma, Jonas Bjork, Massimo Bonini, Matthew S. Dyer, Anna Llanes-Pallas, Davide Bonifazi, Mats Persson, Paolo Samori
DOI: 10.1021/ja9032428
J. Am. Chem. Soc.
16 Sep 2009
Abstract
We study the formation of four supramolecular bicomponent networks based on four linear modules (linkers) bridging melamine via triple hydrogen-bonds. We explore at the nanoscale level the phenomena of polymorphism and phase segregation which rule the generation of highly crystalline nanoporous patterns self-assembled at the solid-liquid interface. The investigated linkers include two systems exposing diuracil groups in the alpha and omega position, naphthalene tetracarboxylic diimide and pyromellitic diimide. In situ scanning tunneling microscopy (STM) investigations revealed that, when blended with melamine, out of the four systems, three are able to form two-dimensional (2D) porous architectures, two of which exhibit highly ordered hexagonal structures, while pyromellitic diimide assembles only into one-dimensional (1D) supramolecular arrays. These bicomponent self-assembled monolayers are used as a test bed to gain detailed insight into phase segregation and polymorphism in 2D supramolecular systems by exploring the contribution of hydrogen-bond energy and periodicity, molecular flexibility, concentration and ratio of the components in solution as well as the effect of annealing via time-dependent and temperature-modulated experiments. These comparative studies, obtained through a joint experimental and computational analysis, offer new insights into strategies toward the bottom-up fabrication of highly ordered tunable nanopatterning at interfaces mediated by hydrogen bonds.
Supramolecular Chemistry at Interfaces: Molecular Recognition on Nanopatterned Porous Surfaces
Davide Bonifazi, Stefan Mohnani, Anna Llanes-Pallas
Journal
Chem.-Eur. J.
Date
07/2009
Chem.-Eur. J.
07/2009
Supramolecular Chemistry at Interfaces: Molecular Recognition on Nanopatterned Porous Surfaces
13 Jul 2009
Abstract
Through the illustration of key examples that have recently appeared in the literature, the intention of this review is to provide a perspective of current advances on the molecular recognition at the interfaces aimed at the engineering of multifunctional organic-based materials. The great interest in such systems has been motivated by the need to fabricate smaller and smaller components in order to improve, for example, the information storage capabilities of classical silicon-based devices. Although great progress has been achieved on the exploitation of top-down approaches, strong hope is now put on the development of hybrid devices in which the elementary components are replaced with single organic molecules. Nevertheless, the drive towards such devices is restricted by both their stability and difficulties to precisely control and manipulate the structural organisation at the molecular level. To overcome these restrictions, the use of nano-templated surfaces featuring porous domains in which responsive functional molecules can be precisely accommodated at the single-molecule level is one of the most promising approaches. In the first part of this manuscript, we therefore illustrate the main engineering strategies [1) through non-covalent interactions, 2) surface-con fined covalent reactions and 3) assembly of pre-organised cavities such as synthetic macrocycles] currently in use to create two-dimensional (2D) patterned surfaces displaying porous structures at the nanoscale level. Such networks, featuring periodic hollow domains (controllable both in shape and size), are of particular significance as their cavities can be used as receptors for the recognition of remotely controllable functional molecules. In the second part, the confinement of molecular guests within the cavities is discussed, emphasising the selectivity and dynamics of key assemblies, with a particular focus on the biomolecular recognition and post-assembly covalent functionalisation, which could provide the opportunity to fabricate devices currently beyond our reach on an unprecedented precision and efficiency. All the examples will be discussed in terms of structural organisation as studied by scanning tunnelling microscopy (STM) techniques.
Organic functionalisation and characterisation of single-walled carbon nanotubes
Prabhpreet Singh, Stephane Campidelli, Silvia Giordani, Davide Bonifazi, Alberto Bianco, Maurizio Prato
DOI: 10.1039/b518111a
Journal
Chem. Soc. Rev.
Date
06/2009
Chem. Soc. Rev.
06/2009
Organic functionalisation and characterisation of single-walled carbon nanotubes
Prabhpreet Singh, Stephane Campidelli, Silvia Giordani, Davide Bonifazi, Alberto Bianco, Maurizio Prato
DOI: 10.1039/b518111a
Chem. Soc. Rev.
22 Jun 2009
Abstract
Since carbon nanotubes (CNTs) display unique structures and remarkable physical properties, a variety of applications have emerged in both materials and life sciences. In terms of applications, the functionalisation of nanotubes is extremely important, as it increases their solubility and processability, and combines the unique properties of single-walled carbon nanotubes (SWCNTs) with those of other classes of materials. A number of methods have been developed, which can be divided into two major approaches: (1) non-covalent supramolecular modifications, and (2) covalent functionalisation. In this tutorial review, we survey the covalent modification of SWCNTs with organic moieties, and illustrate the major analytical techniques routinely used to characterise the functionalised materials.
Photoinduced structural modifications in multicomponent architectures containing azobenzene moieties as photoswitchable cores
Joceline Zeitouny, Claudia Aurisicchio, Davide Bonifazi, Rita De Zorzi, Silvano Geremia, Massimo Bonini, Carlos-Andres Palma, Paolo Samori, Andrea Listorti, Abdelhalim Belbakra, Nicola Armaroli
DOI: 10.1039/b905287a
Journal
J. Mater. Chem.
Date
05/2009
J. Mater. Chem.
05/2009
Photoinduced structural modifications in multicomponent architectures containing azobenzene moieties as photoswitchable cores
Joceline Zeitouny, Claudia Aurisicchio, Davide Bonifazi, Rita De Zorzi, Silvano Geremia, Massimo Bonini, Carlos-Andres Palma, Paolo Samori, Andrea Listorti, Abdelhalim Belbakra, Nicola Armaroli
DOI: 10.1039/b905287a
J. Mater. Chem.
20 May 2009
Abstract
Four novel pi-conjugated chromophores with an azobenzene core (1-4) have been synthesized exploiting Pd-catalysed cross-coupling reactions between ethynyl-bearing azobenzene cores and suitably-designed peripheral groups. While in molecules 2 and 3 the azobenzene core is equipped, respectively, with ethynyl and 1,3-butadiyne spacers terminated with a substituted aniline, molecule 4 is an homologue of derivative 2 in which the terminal moieties are replaced by meso-substituted Zn-porphyrins. X-Ray crystallographic studies of substituted azobenzene 2 reveal a nearly planar arrangement of the four phenyl rings and the trans configuration of the N=N central unit. The UV-Vis absorption spectrum of molecule 1 in cyclohexane (CHX) is very similar to that of unsubstituted azobenzenes; upon irradiation at the maximum of the intense pi-pi absorption feature (360 nm), 1 undergoes trans -> cis photoisomerization reaching a photostationary state. The process is fully reversible both photochemically and thermally (ca. 120 min in the dark). The UV-Vis electronic absorption features of 2-4 are dramatically different compared to those of 1, but the photochemical process can still be traced and exhibits full reversibility in CHX. Also in the case of compound 4, where the photoreactive azobenzene excited states might be quenched by the low-lying porphyrin electronic levels, the photoreaction does occur. Extensive STM investigations of self-assembled monolayers (SAMs) of 2 and 3 at the solid/liquid interface were performed by means of scanning tunneling microscopy (STM) on highly oriented pyrolytic graphite (HOPG). It is evidenced that only the trans isomer can be physisorbed on the surface whereas the cis form, either produced under illumination in situ or prepared by irradiation of the solution prior to deposition (ex-situ), is never observed on the surface. The smallest azobenzene 1 and the bisporphyrin system 4 did not physisorb onto the surface because of the very small size and the bulky 3,5-di(tert-butyl)phenyl groups hindering flat adsorption on HOPG, respectively.
Conformation-controlled networking of H-bonded assemblies on surfaces
Manfred Matena, Anna Llanes-Pallas, Mihaela Enache, Thomas Jung, Johan Wouters, Benoit Champagne, Meike Stoehr, Davide Bonifazi
DOI: 10.1039/b902120e
Journal
Chem. Commun.
Date
05/2009
Chem. Commun.
05/2009
Conformation-controlled networking of H-bonded assemblies on surfaces
Manfred Matena, Anna Llanes-Pallas, Mihaela Enache, Thomas Jung, Johan Wouters, Benoit Champagne, Meike Stoehr, Davide Bonifazi
DOI: 10.1039/b902120e
Chem. Commun.
1 May 2009
Abstract
A temperature-induced phase transition of a 2D H-bonded assembly, enabling quadruple H-bonding interactions, from a hexagonal porous network into a close-packed rhombic arrangement has been observed on Ag(111) by STM imaging.
Selective Formation of Bi-Component Arrays Through H-Bonding of Multivalent Molecular Modules
Luc Piot, Carlos-Andres Palma, Anna Llanes-Pallas, Maurizio Prato, Zsolt Szekrenyes, Katalin Kamaras, Davide Bonifazi, Paolo Samori
Journal
Adv. Funct. Mater.
Date
04/2009
Adv. Funct. Mater.
04/2009
Selective Formation of Bi-Component Arrays Through H-Bonding of Multivalent Molecular Modules
Luc Piot, Carlos-Andres Palma, Anna Llanes-Pallas, Maurizio Prato, Zsolt Szekrenyes, Katalin Kamaras, Davide Bonifazi, Paolo Samori
Adv. Funct. Mater.
23 Apr 2009
Abstract
Here, the formation of discrete supramolecular mono- and bi-component architectures from novel and multivalent molecular modules bearing complementary recognition moieties that are prone to undergo multiple H-bonds, such as 2,6-di(acetylamino)pyridine and uracil residues, is described. These nanostructured H-bonded arrays, including dimeric and pentameric species, are thoroughly characterized in solution by NMR, in the solid state by FT-IR, and at the solid-liquid interface by means of scanning tunneling microscopy. The employed strategy is extremely versatile as it relies on the tuning of the valency, size, and geometry of the molecular modules; thus, it may be of interest for the bottom-up fabrication of nanostructured functional materials with sub-nanometer precision.
Synthesis, photophysical, electrochemical, and electrochemiluminescent properties of 5,15-bis(9-anthracenyl)porphyrin derivatives
Chloe Sooambar, Vincent Troiani, Carlo Bruno, Massimo Marcaccio, Francesco Paolucci, Andrea Listorti, Abdelhalim Belbakra, Nicola Armaroli, Alessandra Magistrato, Rita De Zorzi, Silvano Geremia, Davide Bonifazi
DOI: 10.1039/b820210a
Journal
Org. Biomol. Chem.
Date
04/2009
Org. Biomol. Chem.
04/2009
Synthesis, photophysical, electrochemical, and electrochemiluminescent properties of 5,15-bis(9-anthracenyl)porphyrin derivatives
Chloe Sooambar, Vincent Troiani, Carlo Bruno, Massimo Marcaccio, Francesco Paolucci, Andrea Listorti, Abdelhalim Belbakra, Nicola Armaroli, Alessandra Magistrato, Rita De Zorzi, Silvano Geremia, Davide Bonifazi
DOI: 10.1039/b820210a
Org. Biomol. Chem.
16 Apr 2009
Abstract
Novel 5,15-bis(9-anthracenyl)porphyrin derivatives (1a, 1b) were synthesized by stepwise Suzuki-type coupling reactions using anthracenyl-boronates bearing various electronically active moieties. Absorption spectra of these porphyrin conjugates reveal some degree of delocalisation with the directly linked chromophores, particularly in the case of anthracenyl-porphyrin bearing dimethylanilino moieties at the two extremities. Fluorescence and 77 K phosphorescence properties indicate that the excitation energy is invariably funnelled to the lowest singlet and triplet states of the porphyrin chromophore. The latter levels have been probed also by transient absorption spectroscopy, showing the typical triplet features detected in meso-substituted porphyrins. Extensive electrochemical studies have been performed to unravel the electronic properties of the newly synthesized porphyrins. Low-temperature cyclic voltammetry investigations showed that the anthracenyl-porphyrins are capable of undergoing as many as four electron transfer processes. In particular, by means of UV-Vis-NIR spectroelectrochemical measurements, a NIR-centred intramolecular photoinduced intervalence charge transfer (IV-CT) from a neutral N,N-dimethylanilino moiety to the N, N-dimethylanilino radical cation has been observed for the doubly-oxidised porphyrin 1b(2+). The molecules also showed unexpected electrogenerated chemiluminescence properties, which revealed to be largely controlled by the electronic characteristics of the peripheral anthracenyl substituents. The structural and the electronic properties of these complexes have been also characterised by DFT calculations, as well as by X-ray crystallographic analyses.
Multifunctional hybrid materials composed of [60]fullerene-based functionalized-single-walled carbon nanotubes
Silvia Giordani, Jean-Francois Colomer, Fabrizio Cattaruzza, Jessica Alfonsi, Moreno Meneghetti, Maurizio Prato, Davide Bonifazi
Journal
Carbon
Date
03/2009
Carbon
03/2009
Multifunctional hybrid materials composed of [60]fullerene-based functionalized-single-walled carbon nanotubes
Silvia Giordani, Jean-Francois Colomer, Fabrizio Cattaruzza, Jessica Alfonsi, Moreno Meneghetti, Maurizio Prato, Davide Bonifazi
Carbon
1 Mar 2009
Abstract
We report the synthesis and characterization of several hybrid [60]fullerene-SWCNT materials that combine [60]fullerenes with appended photoactive ferrocenyl or porphyrinyl functionalities and SWCNTs into a single multifunctional structure, where the dyads are covalently attached to the exo-surface of SWCNTs. The structural properties of all hybrids have been characterized using a large variety of spectroscopic and HR-TEM techniques. Raman spectra showed how all SWCNTs were functionalized and the presence of functional groups in the nanotube derivatives. Furthermore, these spectra reveal a new electronic activity of the compounds due to the interaction of the functional groups with the SWCNT frameworks. XPS investigations have documented the presence of [60]fullerene derivatives around the exo-surface of the oxidized SWCNT walls, exhibiting a characteristic photoelectron N 1s emission peak at 400.3 eV. Very importantly, by means of HR-TEM investigations we have also observed the presence of the [60]fullerene functions on the SWCNT outer surface by imaging spherical structures. The presence of the porphyrinyl and ferrocenyl fragments, which can act as effective chromophores and electroactive species, makes this class of materials very interesting for applications in optoelectronics and photovoltaics, and bio-applications, for example in the field of diagnosis and treatment. (C) 2008 Elsevier Ltd. All rights reserved.
Cap removal and shortening of double-walled and very-thin multi-walled carbon nanotubes under mild oxidative conditions
Riccardo Marega, Gianluca Accorsi, Moreno Meneghetti, Andrea Parisini, Maurizio Prato, Davide Bonifazi
Journal
Carbon
Date
03/2009
Carbon
03/2009
Cap removal and shortening of double-walled and very-thin multi-walled carbon nanotubes under mild oxidative conditions
Riccardo Marega, Gianluca Accorsi, Moreno Meneghetti, Andrea Parisini, Maurizio Prato, Davide Bonifazi
Carbon
1 Mar 2009
Abstract
Homogeneous carbon-based materials were prepared for endohedral functionalization starting from carbon nanotubes (CNT) samples mainly containing double-walled and very-thin multi-walled CNTs. Comprehensive structural characterization of end-opened and shortened CNTs, which were prepared under mild oxidative conditions using dilute aqueous H2O2 solutions, is reported. 90% of the 400 measured nanotubes were shorter than 1 mu m with an average value of about 500 nm. TEM observations have confirmed the shortening and opening of the end caps. TGA analysis and Raman spectroscopy showed a reduced impurity content after the oxidation treatment. (C) 2008 Elsevier Ltd. All rights reserved.
Engineering of Supramolecular H-Bonded Nanopolygons via Self-Assembly of Programmed Molecular Modules
Anna Llanes-Pallas, Carlos-Andres Palma, Luc Piot, Abdelhalim Belbakra, Andrea Listorti, Maurizio Prato, Paolo Samori, Nicola Armaroli, Davide Bonifazi
DOI: 10.1021/ja807530m
Journal
J. Am. Chem. Soc.
Date
01/2009
J. Am. Chem. Soc.
01/2009
Engineering of Supramolecular H-Bonded Nanopolygons via Self-Assembly of Programmed Molecular Modules
Anna Llanes-Pallas, Carlos-Andres Palma, Luc Piot, Abdelhalim Belbakra, Andrea Listorti, Maurizio Prato, Paolo Samori, Nicola Armaroli, Davide Bonifazi
DOI: 10.1021/ja807530m
J. Am. Chem. Soc.
21 Jan 2009
Abstract
Discrete and multicomponent nanoscale noncovalent assemblies on surfaces featuring polygonal porous domains are presented. The molecular engineering concept involves multivalent molecular modules that are preprogrammed to undergo heteromolecular recognition by exploiting complementary multiple H bonds. Two types of molecular modules have been engineered: (i) a linear unit of twofold symmetry exposing two 2,6-di(acylamino)pyridyl[donor-acceptor-donor (DAD)] recognition sites at its extremities with a 180 degrees orientation relative to each other and (ii) an angular unit constituted by a 1,3,6,8-tetraethynylpyrene core peripherally functionalized with four uracil groups [acceptor-donor-acceptor (ADA)] positioned at 60 degrees and 120 degrees relative to each other. These molecular modules self-assemble through H-bonds between the complementary recognition sites, forming supramolecular architectures. Their symmetry depends upon the type of each individual subunit and the stoichiometry as well as on the combination and distribution of the main symmetry axes. These so-formed two-dimensional (2D) supramolecular oligomers have been studied in solution by optical spectroscopy and on highly ordered pyrolitic graphite (HOPG) substrates by scanning tunneling microscopy (STM) at the solid-liquid interface. Steady-state UV/vis absorption and emission titration measurements suggest the reversible formation of multiple oligomeric species with slightly modulated fluorescence spectra. This likely reflects the presence of various aggregates between the two polytopic receptors, which exhibit somewhat different electronic delocalization as a function of the aggregate size. The presence of multiple species is further confirmed by time-resolved luminescence measurements: lifetime values are fitted as double/multiple exponentials and are always shorter than 6.5 ns. The formation of several oligomeric species is further supported by in situ STM measurements at the solid-liquid interface that provided evidence, with submolecular resolution, for the formation of multicomponent and discrete 2D polygon-like assemblies. We highlight the role of accurate control of the concentration required to image on the surface the 2D oligomeric species formed in solution, which allows us to bypass the determinant role of the substrate-molecule interactions in forming the thermodynamically stable monocomponent architectures at the solid-liquid interface.
Engineering spherical nanostructures through hydrogen bonds
K. Yoosaf, Abdelhalim Belbakra, Nicola Armaroli, Anna Llanes-Pallas, Davide Bonifazi
DOI: 10.1039/b820462d
Journal
Chem. Commun.
Date
01/2009
Chem. Commun.
01/2009
Engineering spherical nanostructures through hydrogen bonds
K. Yoosaf, Abdelhalim Belbakra, Nicola Armaroli, Anna Llanes-Pallas, Davide Bonifazi
DOI: 10.1039/b820462d
Chem. Commun.
14 Jan 2009
Abstract
Chromophoric acetylenic scaffolds bearing complementary uracyl and 2,6-di(acetylamino) pyridyl moieties undergo supramolecular recognition and generate uniform nanoparticles, as observed by UV-Vis, AFM and TEM measurements.
2008
Title
Trimodular engineering of linear supramolecular miniatures on Ag(111) surfaces controlled by complementary triple hydrogen bonds
Anna Llanes-Pallas, Manfred Matena, Thomas Jung, Maurizio Prato, Meike Stoehr, Davide Bonifazi
Journal
Angew. Chem.-Int. Edit.
Date
09/2008
Angew. Chem.-Int. Edit.
09/2008
Trimodular engineering of linear supramolecular miniatures on Ag(111) surfaces controlled by complementary triple hydrogen bonds
Anna Llanes-Pallas, Manfred Matena, Thomas Jung, Maurizio Prato, Meike Stoehr, Davide Bonifazi
Angew. Chem.-Int. Edit.
22 Sep 2008
Abstract
Simultaneous three-component assembly on surfaces mediated by triple H-bonding interactions leading to the formation of linear supramolecular miniatures has been studied on Ag(111) surfaces by STM. In particular, the complementary assembly of two linear modules (see picture, blue and green) and an anthracenyl-capped molecular stopper (red) led to the formation of discrete linear oligomeric, pentameric, and trimeric nanoassemblies.
Pre-programmed bicomponent porous networks at the solid-liquid interface: the low concentration regime
Carlos-Andres Palma, Massimo Bonini, Anna Llanes-Pallas, Thomas Breiner, Maurizio Prato, Davide Bonifazi, Paolo Samori
DOI: 10.1039/b811534f
Journal
Chem. Commun.
Date
09/2008
Chem. Commun.
09/2008
Pre-programmed bicomponent porous networks at the solid-liquid interface: the low concentration regime
Carlos-Andres Palma, Massimo Bonini, Anna Llanes-Pallas, Thomas Breiner, Maurizio Prato, Davide Bonifazi, Paolo Samori
DOI: 10.1039/b811534f
Chem. Commun.
18 Sep 2008
Abstract
The control over the formation of a bicomponent porous network was attained by self-assembly at the solid-liquid interface, exploiting triple H-bonds between melamine and bis-uracyl modules.
Functionalization of Si(100) with ferrocene derivatives via click chemistry
Andrea G. Marrani, Enrique A. Dalchiele, Robertino Zanoni, Franco Decker, Fabrizio Cattaruzza, Davide Bonifazi, Maurizio Pratoc
Journal
Electrochim. Acta
Date
04/2008
Electrochim. Acta
04/2008
Functionalization of Si(100) with ferrocene derivatives via click chemistry
Andrea G. Marrani, Enrique A. Dalchiele, Robertino Zanoni, Franco Decker, Fabrizio Cattaruzza, Davide Bonifazi, Maurizio Pratoc
Electrochim. Acta
20 Apr 2008
Abstract
We prepared ferrocene-modified silicon surfaces through a three-step procedure consisting of the photochemical anchoring of 11-bromo-1-undecene on H-Si(I 00), followed by treatment with NaN3 and by a reaction with ethynylferrocene via azide-alkyne Huisgen cycloaddition reaction, also known as click chemistry. The advantages of this approach are multiple: the synthetic approach is flexible, provided a C C tethering arm is present on the molecule of interest; a self-assembled hydrocarbon chain can guarantee a good coverage and resistance to the further synthetic steps; the redox centers are located at the outer surface, where a good contact with the electrolyte becomes possible. We have monitored the progression of the reaction steps by XPS, and characterized the resulting new hybrid on Si by electrochemical methods. The presence and chemical nature of the redox species covalently attached to the SAM on Si has been evaluated by XPS, while the overall coverage has been calculated by CV measurements. A reversible electrochemical response has been evidenced for the hybrids and the progressive ageing followed at thousands of oxidation-reduction cycles. (c) 2007 Elsevier Ltd. All rights reserved.
Redox-active Si(100) surfaces covalently functionalised with [60]fullerene conjugates: new hybrid materials for molecular-based devices
Fabrizio Cattaruzza, Anna Llanes-Pallas, Andrea G. Marrani, Enrique A. Dalchiele, Franco Decker, Robertino Zanoni, Maurizio Prato, Davide Bonifazi
DOI: 10.1039/b717438a
Journal
J. Mater. Chem.
Date
02/2008
J. Mater. Chem.
02/2008
Redox-active Si(100) surfaces covalently functionalised with [60]fullerene conjugates: new hybrid materials for molecular-based devices
Fabrizio Cattaruzza, Anna Llanes-Pallas, Andrea G. Marrani, Enrique A. Dalchiele, Franco Decker, Robertino Zanoni, Maurizio Prato, Davide Bonifazi
DOI: 10.1039/b717438a
J. Mater. Chem.
1 Feb 2008
Abstract
Herein, we report the covalent immobilisation, through Si-C bonds, of various [60]fullerene derivatives on flat silicon surfaces following three different preparative protocols. Each synthetic strategy comprises a two-step approach that includes a pre-modification step of the Si(100) surface with an organic monolayer bearing a terminal functionality that undergoes a bond-forming reaction with a [60]fullerene synthon as characterized by X-ray photoelectron spectroscopy (XPS) measurements. Water contact angle measurements clearly showed a characteristic change of the surface hydrophobicity upon covalent immobilisation of the carbon functions. The hybrid [4b-Si(100)] surfaces, containing [60]fullerene-ferrocene fragments, were also investigated by means of cyclic voltammetry (CV), and were revealed to be exceptionally robust towards repeated reduction-oxidation cycles. Moreover, several surface-confined redox couples were observed in CH3CN solution. The surface coverage was measured to be ca. 2.5 x 10(-11) mol cm(-2).
2007
Title
Wet adsorption of a luminescent EuIII complex on carbon nanotubes sidewalls
Gianluca Accorsi, Nicola Armaroli, Andrea Parisini, Moreno Meneghetti, Riccardo Marega, Maurizio Prato, Davide Bonifazi
Journal
Adv. Funct. Mater.
Date
10/2007
Adv. Funct. Mater.
10/2007
Wet adsorption of a luminescent EuIII complex on carbon nanotubes sidewalls
Gianluca Accorsi, Nicola Armaroli, Andrea Parisini, Moreno Meneghetti, Riccardo Marega, Maurizio Prato, Davide Bonifazi
Adv. Funct. Mater.
15 Oct 2007
Abstract
A Eu-III complex, tris-dibenzoylmethane mono-1,10-phenanthroline-europium(III) [Eu(DBM)(3)(Phen)], can be easily adsorbed If in situ via hydrophobic interactions to single-walled carbon nanotube (SWNT) surfaces from a methanol solution. The Eu-III containing material has been comprehensively characterized via thermogravimetric analysis (TGA), UV-vis-NIR absorption and luminescence spectroscopy, Raman spectroscopy, atomic force microscopy (AFM), high-resolution transmission electron microscopy (HR-TEM)), Z-contrast scanning transmission electron microscopy (STEM) imaging, and energy dispersive X-ray spectroscopy (EDS). The photophysical investigations revealed that the presence of a SWNT framework does not affect the lanthanide-centered luminescence stemming from the characteristic electronic transitions within the 4f shell of the Eu-III, ions. Such straightforward synthetic route leads to the preparation of luminescent SWNTs without significantly affecting the electronic and structural properties of the carbon framework, opening new possibilities of designing new classes of CNTs for biomedical applications.
A supramolecular multiposition rotary device
Nikolai Wintjes, Davide Bonifazi, Fuyong Cheng, Andreas Kiebele, Meike Stoehr, Thomas Jung, Hannes Spillmann, Francois Diederich
Journal
Angew. Chem.-Int. Edit.
Date
05/2007
Angew. Chem.-Int. Edit.
05/2007
A supramolecular multiposition rotary device
Nikolai Wintjes, Davide Bonifazi, Fuyong Cheng, Andreas Kiebele, Meike Stoehr, Thomas Jung, Hannes Spillmann, Francois Diederich
Angew. Chem.-Int. Edit.
16 May 2007
Abstract
A supramolecular rotary device, reminiscent of a mechanical rotary switch, was engineered by a bottom-up approach. Self-assembly of a functionalized porphyrin molecule leads to the formation of a porous network that features chiral cavities. These serve as hosts for molecular guests, which can be induced to rotate either thermally or by using the scanning tunneling microscopy tip (see images).
Nonlinear optical properties of ferrocene- and porphyrin-[60]fullerene dyads
Evangelia Xenogiannopoulou, Miroslav Medved, Kostas Iliopoulos, Stelios Couris, Manthos G. Papadopoulos, Davide Bonifazi, Chloe Sooambar, Aurelio Mateo-Alonso, Maurizio Prato
Journal
ChemPhysChem
Date
05/2007
ChemPhysChem
05/2007
Nonlinear optical properties of ferrocene- and porphyrin-[60]fullerene dyads
Evangelia Xenogiannopoulou, Miroslav Medved, Kostas Iliopoulos, Stelios Couris, Manthos G. Papadopoulos, Davide Bonifazi, Chloe Sooambar, Aurelio Mateo-Alonso, Maurizio Prato
ChemPhysChem
14 May 2007
Abstract
A series of novel [60]fullerene-ferrocene and [60]fullerene-porphyrin dyads, in which a fullerene and an electron donating moiety are attached through a flexible triethylene glycol linker are synthesized and their nonlinear optical (NLO) response studied. Specifically the third-order susceptibility chi((3)) of all fullerene derivatives are measured in toluene solutions by the optical Kerr effect (OKE) technique using 532 nm, 35 ps laser pulses and their second hyperpolarizability gamma are determined. All fullerene dyads studied exhibit enhancement of their NLO response compared to pristine fullerenes which has been attributed to the formation of a charge separated state. All experimentally measured hyperpolarizability gamma values are also calculated by the semiempirical methods AM1 and PM3. A good correlation is found between the theoretical and experimental values, suggesting that simple semi-empirical methods can be employed for the designing and optimization of the fullerene-containing dyads displaying improved nonlinear responses.
Supramolecular nanostructuring of silver surfaces via self-assembly of [60]fullerene and porphyrin modules
Davide Bonifazi, Andreas Kiebele, Meike Stoehr, Fuyong Cheng, Thomas Jung, Francois Diederich, Hannes Spillmann
Journal
Adv. Funct. Mater.
Date
05/2007
Adv. Funct. Mater.
05/2007
Supramolecular nanostructuring of silver surfaces via self-assembly of [60]fullerene and porphyrin modules
Davide Bonifazi, Andreas Kiebele, Meike Stoehr, Fuyong Cheng, Thomas Jung, Francois Diederich, Hannes Spillmann
Adv. Funct. Mater.
7 May 2007
Abstract
Recent achievements in our laboratory toward the bottom-up fabrication of addressable multicomponent molecular entities obtained by self-assembly of C-60 and porphyrins on Ag(100) and Ag(111) surfaces are described.. Scanning tunneling microscopy (STM) studies on ad-layers constituting monomeric and triply linked porphyrin modules showed that the molecule:, self-organize into ordered supramolecular assemblies, the ordering of which is controlled by the porphyrin chemical structure, the metal substrate, and the surface coverage. Specifically, the successful preparation of unprecedented two-dimensional porphyrin-based assemblies featuring regular pores on Ag(111) surfaces has been achieved. Subsequent co-deposition of C-60 molecules on top of the porphyrin monolayers results in selective self-organization into ordered molecular hybrid bilayers, the organization of which is driven by both fullerene coverage and porphyrin structure. In all-ordered fullerene-porphyrin assemblies, the C-60 guests organize, unusually, into long chains and/or two-dimensional arrays. Furthermore, sublimation of C-60 on top of the porous porphyrin network reveals the selective long-range inclusion of the fullerene guests within the hosting cavities. The observed mode of the C-60 self-assembly originates from a delicate equilibrium between substrate-molecule and molecule-molecule interactions involving charge-transfer processes and conformational reorganizations as a consequence of the structural adaptation, of the fullerene-porphyrin bilayer.
2006
Title
Supramolecular [60]fullerene chemistry on surfaces
Davide Bonifazi, Olivier Enger, Francois Diederich
DOI: 10.1039/b604308a
Journal
Chem. Soc. Rev.
Date
12/2006
Chem. Soc. Rev.
12/2006
Supramolecular [60]fullerene chemistry on surfaces
19 Dec 2006
Abstract
This critical review documents the exceptional range of research avenues in [60]fullerene-based monolayers showing unique and spectacular physicochemical properties which prompted such materials to have potential applications in several directions, ranging from sensors and photovoltaic cells to nanostructured devices for advanced electronic applications, that have been pursued during the past decade. It illustrates how progress in covalent [60]fullerene functionalisation led to the development of spectacular surface-immobilised architectures, including dyads and triads for photoinduced electron and energy transfer, self-assembled on a wide variety of surfaces. All of these molecular assemblies and supramolecular arrays feature distinct properties as a consequence of the presence of different molecular units and their spatial arrangement. Since the properties of [60]fullerene-containing films are profoundly controlled by the deposition conditions, substrate of adsorption, and influenced by impurities or disordered surface structures, the progress of such new [60]fullerene-based materials strongly relies on the development of new versatile and broad preparative methodologies. Therefore, the systematic exploration of the most common approaches to prepare and characterise [60]fullerene-containing monolayers embedded into two- or three-dimensional networks will be reviewed in great detail together with their main limitations. Recent investigations hinting at potential technological applications addressing many important fundamental issues, such as a better understanding of interfacial electron transfer, ion transport in thin films, photovoltaic devices and the dynamics associated with monolayer self-assembly, are also highlighted.
Adsorption and dynamics of long-range interacting fullerenes in a flexible, two-dimensional, nanoporous porphyrin network
Andreas Kiebele, Davide Bonifazi, Fuyong Cheng, Meike Stoehr, Francois Diederich, Thomas Jung, Hannes Spillmann
Journal
ChemPhysChem
Date
07/2006
ChemPhysChem
07/2006
Adsorption and dynamics of long-range interacting fullerenes in a flexible, two-dimensional, nanoporous porphyrin network
Andreas Kiebele, Davide Bonifazi, Fuyong Cheng, Meike Stoehr, Francois Diederich, Thomas Jung, Hannes Spillmann
ChemPhysChem
17 Jul 2006
Abstract
Herein, a detailed investigation of the adsorption and dynamics of C-60 and C-70 fullerenes hosted in a self-assembled, two-dimensional, nanoporous porphyrin network on a solid Ag surface is presented. Time-resolved scanning tunneling microscopy (STM) studies of these supramolecular systems at the molecular scale reveal distinct host-guest interactions giving rise to a pronounced dissimilar mobility of the two fullerenes within the porphyrin network. Furthermore, long range coverage-dependant interactions between the all-carbon guests, which clearly affect their mobility and are likely mediated by a complex mechanism involving the Ag substrate and the flexible porphyrin host network, are observed. At increased fullerene coverage, this unprecedented interplay results in the formation of large fullerene chains and islands. By applying a lattice gas model with nearest-neighbour interactions and by evaluating the fullerence-pair distribution functions, the respective coverage-dependant guest-guest interaction energies are estimated.
Microscopic and spectroscopic characterization of paintbrush-like single-walled carbon nanotubes
Davide Bonifazi, Christophe Nacci, Riccardo Marega, Stephane Campidelli, Gustavo Ceballos, Silvio Modesti, Moreno Meneghetti, Maurizio Prato
DOI: 10.1021/nl060394d
Journal
Nano Lett.
Date
07/2006
Nano Lett.
07/2006
Microscopic and spectroscopic characterization of paintbrush-like single-walled carbon nanotubes
Davide Bonifazi, Christophe Nacci, Riccardo Marega, Stephane Campidelli, Gustavo Ceballos, Silvio Modesti, Moreno Meneghetti, Maurizio Prato
DOI: 10.1021/nl060394d
Nano Lett.
12 Jul 2006
Abstract
Understanding and controlling the chemical reactivity of carbon nanotubes (CNTs) is a fundamental requisite to prepare novel nanoscopic structures with practical uses in materials applications. Here, we present a comprehensive microscopic and spectroscopic characterization of carbon nanotubes which have been chemically modified. Specifically, scanning tunneling microscopy ( STM) investigations of short-oxidized single-walled carbon nanotubes (SWNTs) functionalized with aliphatic chains via amide reaction reveal the presence of bright lumps both on the sidewalls and at the tips. The functionalization pattern is consistent with the oxidation reaction which mainly occurs at the nanotube tips. Thermogravimetric analysis (TGA), steady-state electronic absorption (UV-vis-NIR), and Raman spectroscopic studies confirm the STM observations.
Functionalized carbon nanotubes are non-cytotoxic and preserve the functionality of primary immune cells
Helene Dumortier, Stephanie Lacotte, Giorgia Pastorin, Riccardo Marega, Wei Wu, Davide Bonifazi, Jean-Paul Briand, Maurizio Prato, Sylviane Muller, Alberto Bianco
DOI: 10.1021/nl061160x
Journal
Nano Lett.
Date
07/2006
Nano Lett.
07/2006
Functionalized carbon nanotubes are non-cytotoxic and preserve the functionality of primary immune cells
Helene Dumortier, Stephanie Lacotte, Giorgia Pastorin, Riccardo Marega, Wei Wu, Davide Bonifazi, Jean-Paul Briand, Maurizio Prato, Sylviane Muller, Alberto Bianco
DOI: 10.1021/nl061160x
Nano Lett.
12 Jul 2006
Abstract
Carbon nanotubes are emerging as innovative tools in nanobiotechnology. However, their toxic effects on environment and health have become an issue of strong concern. In the present study, we address the impact of functionalized carbon nanotubes (f-CNTs) on cells of the immune system. We have prepared two types of f-CNTs, following the 1,3-dipolar cycloaddition reaction ( f-CNTs 1 and 2) and the oxidation/ amidation treatment (f-CNTs 3 and 4), respectively. We have found that both types of f-CNTs are uptaken by B and T lymphocytes as well as macrophages in vitro, without affecting cell viability. Subsequently, the functionality of the different cells was analyzed carefully. We discovered that f-CNT 1, which is highly water soluble, did not influence the functional activity of immunoregulatory cells. f-CNT 3, which instead possesses reduced solubility and forms mainly stable water suspensions, preserved lymphocytes functionality while provoking secretion of proinflammatory cytokines by macrophages.