Research Topics
The research activities in the Bonifazi group are centred on ambitious targeted organic synthesis, carbon-based nanostructure chemistry, self-assembly and self-organisation studies to develop practical access to functional organic architectures displaying tailored properties for real world-technologies. Current research interests can be divided in four main topics:
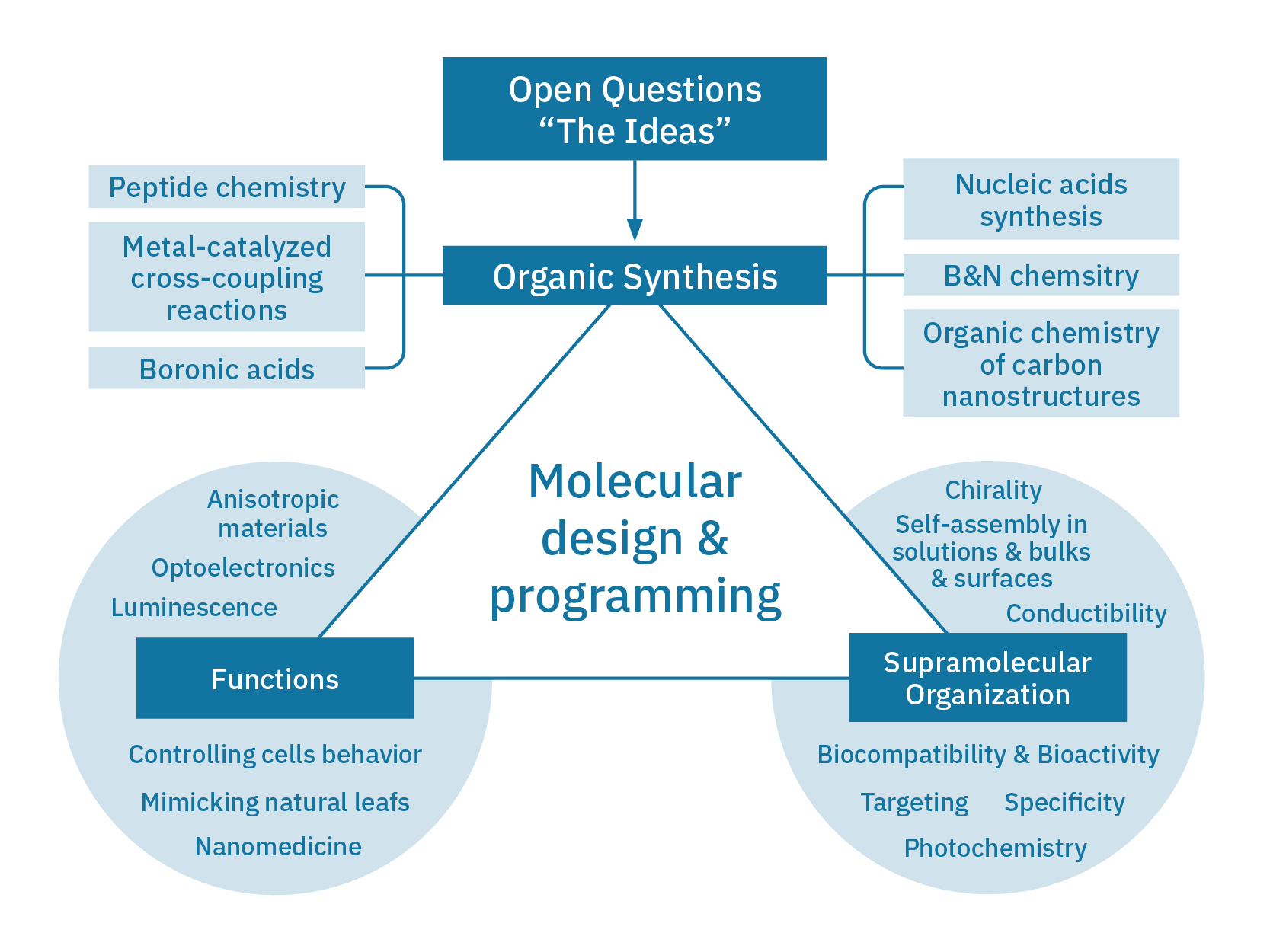
Building on synthetic methods developed by the group for constructing oxygen-embedded PAHs (O-PAHs) via oxidative Pummerer-type annulation (ACIE 2016, 55, 5947; CEJ 2017, 23, 2363; ACIE 2018, 57, 8942; OL 2020, 22, 4283) using Cu(I), we prepared a diverse range of O-PAHs. We isolated their cationic organic crystals through electrocrystallization, revealing pronounced electron delocalization within π-stacked columns in the solid state. Electrical measurements confirmed semiconducting behavior with high charge mobilities, underscoring the potential of heteroatom-doping to engineer semiconducting materials (ACIE 2020, 59, 4106). Another significant advance was the design of N-atom doping patterns to control the optical bandgap of peri-xanthenoxanthene (PXX) derivatives (Chem. Sci. 2022, 13, 6335). Using TD-DFT calculations, we developed a tool to map charge redistribution in singlet-state excitations, identifying atomic sites for N-atom substitution to tailor UV-vis absorption and emission envelopes. Experimental validation involved synthesizing N-doped PXX isomers with distinct substitution patterns, which exhibited blueshifts or redshifts in electronic transitions and demonstrated strong correlations with the predicted data. This work lays the foundation for a new research program,”Chromophore à la Carte”, focused on knowledge-based design of chromophores for light-related applications (e.g., harvesting).
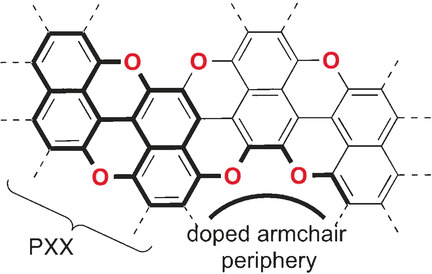
In parallel, we developed one of the first Lewis-Acid-free amino-directed borylation protocols, designing the first series of regularly BN-doped molecular ribbons (JACS 2022, 144, 21470). We systematically prepared zig-zag edged nanoribbons that exhibited progressive bathochromic shifts in their absorption and emission spectra upon extending the polyaromatic framework, high fluorescence quantum yields (up to 86%), and surprising room temperature thermally activated delayed fluorescence (TADF) properties. The distortion of the π-conjugated core was identified as the primary factor promoting spin-orbit coupling, triggering TADF. Building on this, we prepared a series of BN-doped molecules that progressively exhibit marked distortions and TADF properties (in preparation). Pyrene-functionalized derivatives were also developed using this approach and showed thermally activated emission in LEC devices (Adv. Funct. Mater. 2025, just accepted). Simultaneously, to boost the chemical and physical resilience of graphenoids for device applications (Adv. Funct. Mater. 2020, 30, 1906830; ACS Appl. Mater. Interfaces, 2020, 12, 28426), we peripherally functionalized PAHs with unprecedented NBNBN- and OBOBN motifs (submitted), exhibiting bright fluorescence (up to 0.99 quantum yield) and notable resilience to irradiation.
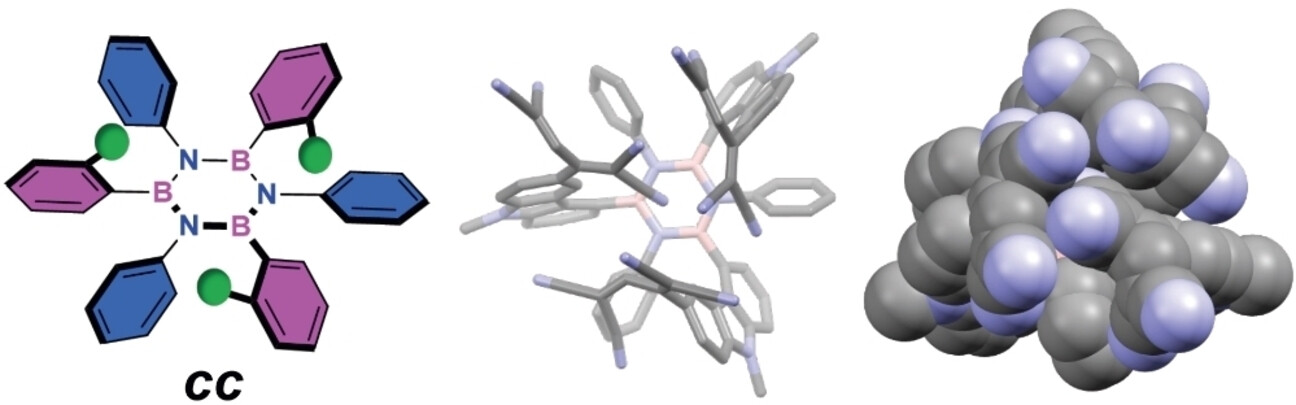
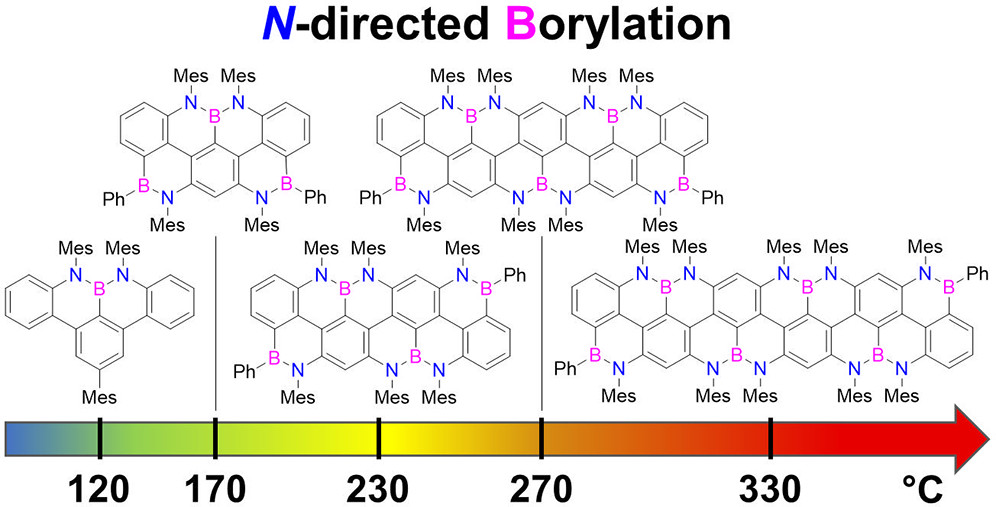
Following our group’s investigations on chemically resilient borazine derivatives by exploiting stereoelectronic effects (Chem. Commun. 2025, 61, 1200) and extend the chemical space of graphenoids in three dimensions, we developed the first diastereoselective synthesis of borazine frameworks. We synthesized propeller-shaped hexaphenylborazines (ACIE 2024, e202416700), using tailored aryl lithium nucleophiles for stereochemical control in borazine substitution reactions. Systematic analysis showed that ortho-substituents at the B-aryl moieties can shield one side of the borazine ring, allowing formation of all-cis (cc) stereoisomers. The resulting borazines were used to create multichromophoric architectures, opening pathways to engineer complex 3D sp²-hybridized structures, such as polymers developed for the preparation of solid-state boron-nitride-containing electrolyte gels for batteries (Chem. Mater. 2022, 34, 10670). One derivative was identified as an efficient photosensitizer for dye-sensitized solar cells (Adv. Energ. Sust. Res. 2025, 2400344). Moreover, we developed thiol-functionalized borazine derivatives that, upon Cu-surface anchoring, were graphitized to prepare BNC-type graphenoid multilayers suitable as conductive electrodes in solar cells (ACS Appl. Mater. Interfaces 2025, 17, 23062). Covalent, borazine-based porous monolayered BN-containing networks were also developed with our collaborators at TUM (two papers in preparation).
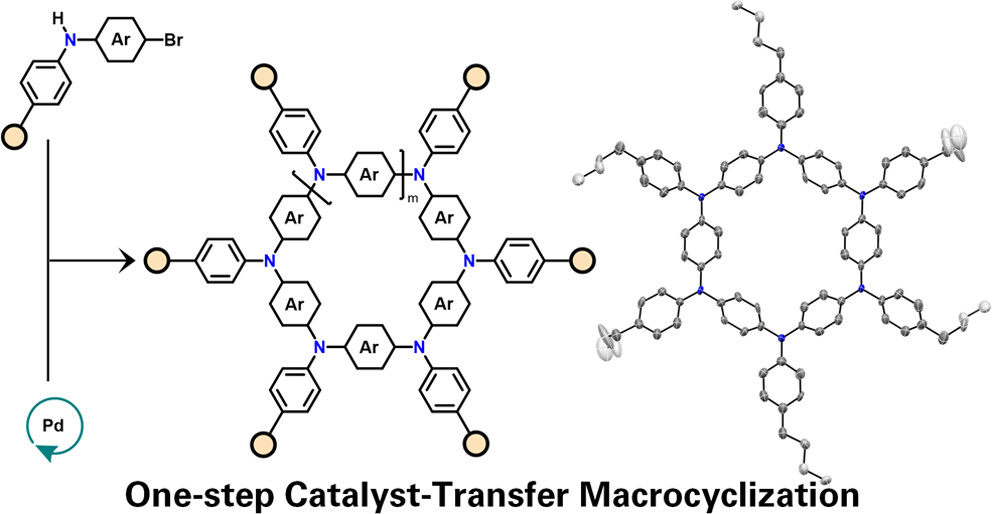
Finally, aiming to mimic natural antennas with multichromophoric cyclic architectures exhibiting strong excitonic coupling, we developed the first example of a one-step catalyst-transfer macrocyclization (CTM) protocol (JACS 2024, 146, 16440), which leverages the Buchwald-Hartwig cross-coupling reaction, featuring a proposed “living” ring-walking catalyst-transfer mechanism that directs the formation of aza[1n]paracyclophanes (ATPs) at mild temperatures in short times, achieving yields over 75%. This accessible synthetic method (JACS Au 2025, 5, 1482) functions effectively under both diluted and concentrated conditions, unlike typical macrocyclization reactions, enabling access to a wide variety of controlled systems.
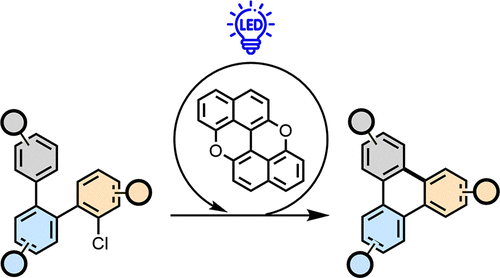
Another pillar of our research has been the development of new radical-based, metal-free methodologies for synthesizing PAHs. Building on our earlier work showing that cost-effective PXX can trigger photoinduced electron transfer (PET) to electron-poor aromatics and generate aryl radicals (CEJ 2018, 24, 4382), we further explored PXX’s versatility as a reducing organic photocatalyst. We demonstrated that PXX enables efficient reduction of halides and coupling to create new C–C bonds (Adv. Synth. Catal. 2021, 363, 4740), including β-arylation of cyclic ketones and Ni-catalyzed cross-couplings. Additionally, we developed a mild, metal-free annulation reaction to synthesize peripherally substituted PAHs from ortho-oligoarylenyl precursors under visible light irradiation (JACS Au 2023, 3, 3045). This approach directly produced triphenylene and other extended C(sp2)-frameworks, including heteroatom-bearing derivatives, with exceptional functional group tolerance. We also discovered that selective photocatalyzed Birch-type reduction of acenes can utilize PXX in high yields (CEJ 2023, 29, e202302129). In these PXX-photocatalyzed systems, radical formation occurs primarily for electron-deficient substrates. To expand the radical approach to electron-rich and unsubstituted aromatics, we developed a modular electrophotocatalytic platform based on phosphonium- and sulfone-bearing PXX congeners, accessing highly reducing excited states (up to –3.44 V vs SCE) via electrochemical reduction and visible-light excitation (submitted). This enabled activation of challenging substrates, such as aryl fluorides and electron-rich alkynes, under mild, metal-free conditions to create C(sp2)–C(sp2) and C(sp)–C(sp2) bonds.
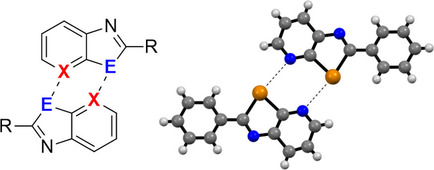
I have extensively explored chalcogen-bonding interactions as a powerful tool (Coord. Chem. Rev. 2020, 423, 213243) for supramolecular engineering of materials at the solid state (new review submitted). Building on our previously developed chalcogenazolo-pyridine scaffold (CGP) that encoded dimeric-like recognition behavior in the solid state (CEJ 2018, 24, 5439), we showed that complex supramolecular polymers featuring head-to-tail halogen and chalcogen-bonded interactions could be obtained (CEJ 2020, 26, 2904) as well as different molecular recognition modes (Cryst. Growth Des 2021, 21, 536) when different substituents were used. Also, we have shown that the CGP-driven recognition perfectly works on surfaces (JACS Au 2024, 4, 2115), as demonstrated by HR-STM imaging and quantum chemical calculations, which showed that PAH-bearing CGP moieties also undergo dimerization on Au(111). In an evolved investigation, we prepared a ditellurophene system bearing a pyridyl unit to establish dimeric interactions. The molecule was found to undergo supramolecular nanoribbons formation at the solid state (ACIE 2022, 61, e202202137). When used as the transport layer, these materials exhibited exceptional hole transport in thin films, demonstrating outstanding performance in LECs and enhancing their durability by approximately 40 hours. This represented the first demonstration of utilizing such interactions to design functional electronic materials. Additionally, we engineered a pyrselen-based recognition motif that consistently dimerizes in solution through ChBIs, providing a framework for a dynamic and directional self-assembly motif that can also develop in the liquid phase (CEJ 2020, 26, 2904).
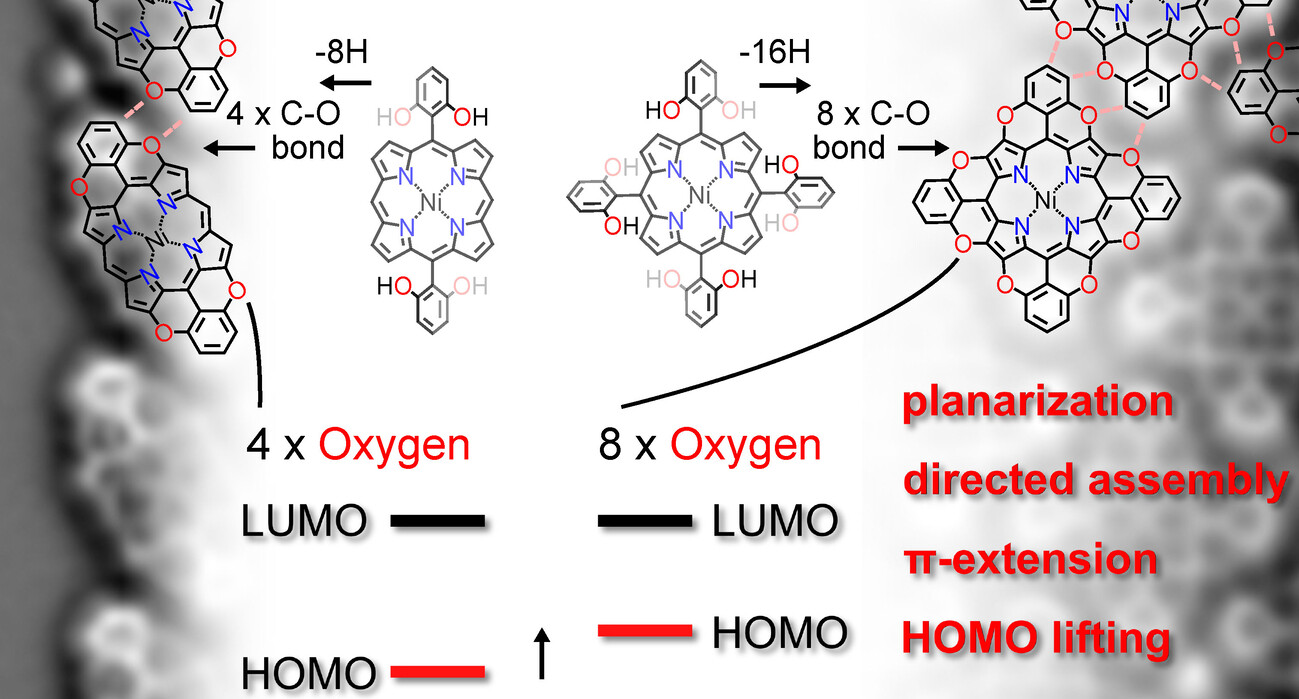
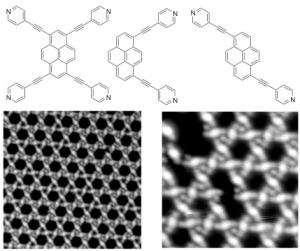
Building on our long-standing research interest in surface-confined supramolecular assemblies and their reactivity, we have recently employed a halogenated PXX-based linker to construct on-surface halogen-bonded networks. High-resolution scanning tunneling microscopy (HR-STM) investigations revealed that the brominated PXX modules undergo a Kagome-type self-assembly on Au(111), stabilized by halogen bonding rather than hydrogen bonding (Nat. Commun. 2020, 11, 2123). Additionally, we established that these molecules can undergo surface-induced polymerization, leading to the formation of O-doped nanoribbons (in preparation). In co-assembly experiments with TCNQ-type acceptor molecules, we observed unprecedented surface-mediated charge transfer accompanied by unique supramolecular arrangements. Extending this line of research, we successfully adapted the Pummerer rearrangement protocol to the surface environment. We developed the first example of on-surface C–O bond formation, yielding O-annulated porphyrins (ACIE 2025, 64, 202412978). These systems form well-defined chiral assemblies governed by C–H···O interactions and exhibit tunable HOMO–LUMO gaps as a function of the number of O-annulations.
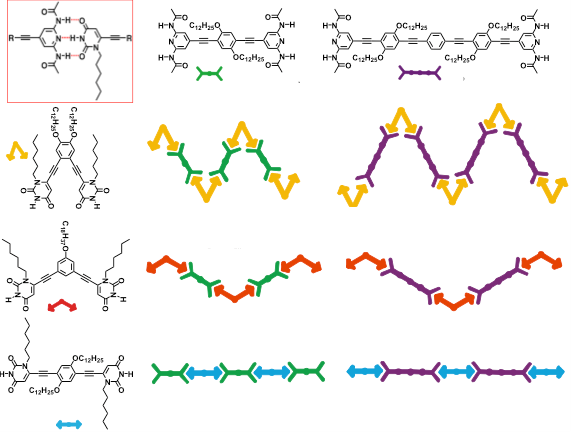
Since its creation at the end of 2006, our team has been very active in the field of supramolecular organic chemistry at interfaces, with the objective of taking advantage of the supramolecular approach as a route to build defined two- and three-dimensional nano-patterned organic networks. We have validated the concept for which complementary H-bonding interactions can serve as powerful and versatile tool for tuning the shape of materials giving access to an enormous number of structurally-organized architectures, mastering their organization from the nanoscale up to the macroscopic level [Chem. Eur. J., 2009, 15, 7004; Coord. Chem. Rev., 2010, 254, 2342; Pure Appl. Chem., 2010, 10, 917; Nanoscale, 2013, 5, 8837]. In a collaborative research program with the group of Prof. M. Stohr (University of Groningen, Holland), exploiting the triply H-bonded uracil-2,6-diamidopyridine recognition motif linked to oligo(phenylene-ethynylene) molecular wires, we have reported the first example, at that time, of a simultaneous three-components H-bonded assembly on Ag(111) surfaces by STM analysis under ultrahigh vacuum conditions (UHV). [Angew. Chem. Int. Ed., 2008, 47, 7726]
Simultaneously, for the first time, we also reported the first writable organic-based surfaces based on thermally switchable 2D porous assembly [Chem. Commun., 2009, 3525]. Discrete assemblies, featuring hosting properties towards hydrophobic porphyrin guests, have been also formed using complementary angular modules. The multicomponent non-covalent H-bonded hollowed assemblies featuring polygonal porous domains (in coll. with Prof. P Samori at Universisty of Strasbourg, France) have also been comprehensively investigated at the solid-liquid interfaces, for the first time showing that both the network and the phase depend on the molecular geometry, concentrations and the peculiar chemical structure [Chem. Commun., 2008, 5289; Adv. Funct. Mater., 2009, 19, 1207; J. Am. Chem. Soc., 2009, 131, 509; J. Am. Chem. Soc., 2009, 131, 13062]. Passing from the molecular to the nanoscale, our team has also shown how the use of H-bonding allowed the tuning of the size and shape of uniform architectures enabling a morphological change to occur from nanoparticle and vesicles [Chem. Commun., 2009, 2830]. The formation of helicoidally organized rods, crater-, prism- and crown-like morphologies have been also fully described [Chem. Eur. J., 2010, 17, 3262; Langmuir, 2011, 27, 1513].
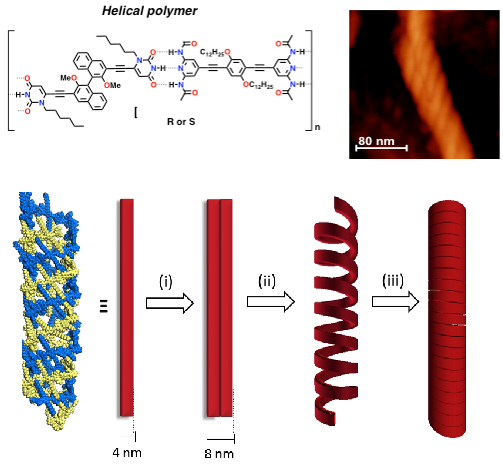
Aiming at introducing asymmetric properties and relate molecular and macroscopic chirality [ChemPlusChem, 2014, 79, 895], we have very recently engineered a new family of BINOLs derivatives undergoing solvent-dependent nanostructuration. Depending on the solvophobic properties of the liquid media, the material can be moulded into spherical, rod-like, fibrous and helical morphologies [J. Am. Chem. Soc., 2015, 137, 8150]. This behaviour is interpreted as a consequence of an interplay between the degree of association of the H-bond recognition, the vapour pressure of the solvent and the solvophobic/solvophilic character of the supramolecular adducts in the different solutions under dynamic conditions, namely during solvent evaporation conditions at room temperature.
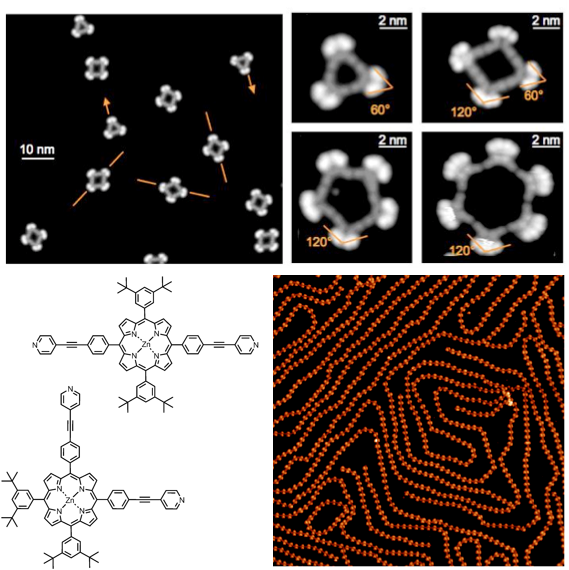
In collaboration with the group of Prof. J. Barth and Prof. W. Auwarter (TUM), we have described for the first time the formation of discrete, tunable and hollowed cyclic supramolecular architectures on insulating boron-nitrate [J. Am. Chem. Soc., 2015, 137, 2420] and on metal surfaces exploiting metal-coordination bonding interactions with pyridyl-bearing ligands [Nano Lett., 2010, 10, 122; J. Am. Chem. Soc., 2010, 132, 6783; ACS Nano, 2010, 6, 4936], the structure of which could be tailored by the geometry, conformational flexibility and functionality of the porphyrin molecular modules. Recently, we have also reported the first patterning of lanthanide and transition metal atoms on surfaces exploiting the orthogonal coordination properties of tetrapyridyl porphyrins [Angew. Chem. Int. Ed., 2015, 54, 6163]. Expanding the topic, a series of differently substituted pyridyl-bearing pyrenyl modules [ACS Nano, 2016, 10, 7665] have been prepared, all showing a different self-assembly behavior (e.g., spangling, crocheting and knitting) depending on the spatial disposition of the pyridyl groups. Extended porous networks showing inherent flexibility and adaptability properties have been prepared exploiting competing non-covalent interactions, using tripodal modules like 1,3,4-tris-pyridyl-benzene [ACS Nano, 2012, 6, 4258; Chem. Eur. J., 2013, 14143]. In a parallel study, in collaboration with the groups of Prof. G. Costantini (Warwick University, United Kingdom) and Prof. A. De Vita (King’s College, United Kingdom), we demonstrated that polyaromatic hydrocarbons form nanostructures at the interface that are ruled by an interplay between van der Waals attractions and intermolecular repulsions, the latter driven by reversible molecule-substrate charge transfer [ACS Nano, 2014, 8, 12356, Nanoscale, 2016, 8, 19004].
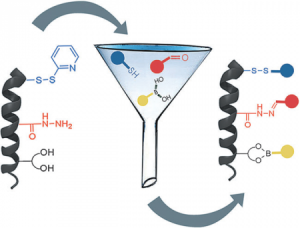
Aiming at introducing a certain functionality on porous self-assembled organic networks, we have very recently initiated a novel research project (sponsored by the ERC Starting Grant 2012, COLORLANDS) in which, taking advantage of the molecular self-assembly, we seek to create a new generation of periodically-organized organic architectures that can structurally control the arrangement of molecular chromophores and/or fluorophores. The ultimate aim is to create a library of supramolecularly-assembled architectures emitting or adsorbing light throughout the visible region that, depending on their spatial organization, are suitable for producing versatile organic materials for device implementation (e.g. an OLED with luminophores or an artificial “leafs” mimicking natural antenna if hosting chromophores). In this respect, we conceived a facile and versatile protocol for the simultaneous use of three orthogonal dynamic covalent reactions, namely disulfide, boronate and acyl hydrazine formation. Using a pre-programmed a-helix peptide, we conceived a facile and versatile protocol to spatially organize tailored blue, red and yellow chromophores [Angew. Chem. Int. Ed., 2015, 54, 15739].